

Genotype-phenotype correlation of gangliosidosis mutations using in silico tools and homology modeling
- PMID: 31367523
- PMCID: PMC6646740
- DOI: 10.1016/j.ymgmr.2019.100495
Genotype-phenotype correlation of gangliosidosis mutations using in silico tools and homology modeling
Abstract
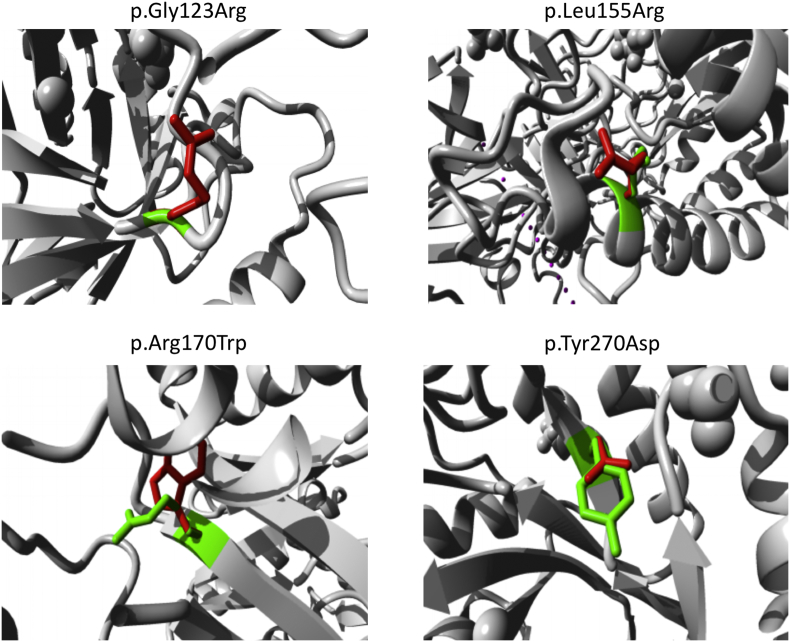
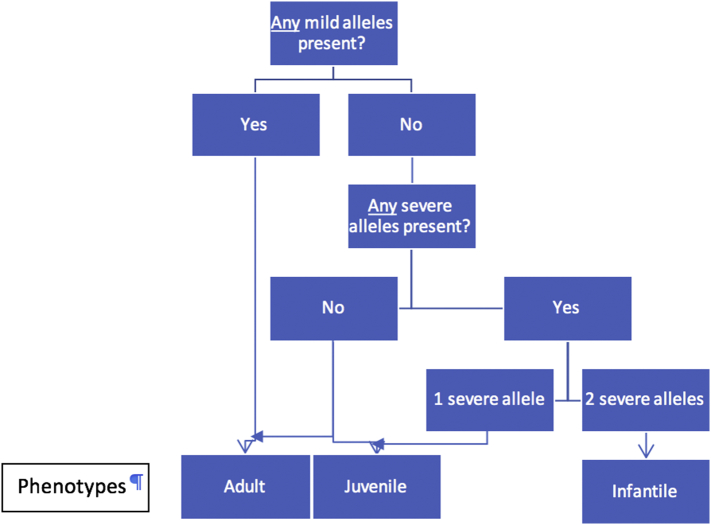
Gangliosidoses, including GM1-gangliosidosis and GM2-gangliosidosis (Tay-Sachs disease and Sandhoff disease), are lysosomal disorders resulting from enzyme deficiencies and accumulation of gangliosides. Phenotypes of gangliosidoses range from infantile, late-infantile, juvenile, and to the adult form. The genotype-phenotype correlation is essential for prognosis and clinical care planning for patients with a gangliosidosis condition. Previously, we have developed a method to establish the genotype-phenotype correlation of another lysosomal disease, mucopolysaccharidosis type I, with in silico tools. This same method was applied to analyze the genotype and phenotype of 38 patients diagnosed with a gangliosidosis disease in the United States. Out of 40 mutations identified, 3 were novel, including p.Tyr192His and p.Phe556Ser of the GLB1 gene and p.Gly461Val of the HEXA gene. Furthermore, the mutant protein structure of all missense mutations was constructed by homology modeling. A systemic structural analysis of these models revealed the specific mechanisms of how each mutation may lead to the disease. In summary, the method developed in this study holds promise as a tool that can be broadly applicable to other lysosomal diseases and monogenic diseases.
Keywords: Disease subtype; Gangliosidosis; Genotype-phenotype correlation; In silico; Lysosomal disorder.
Figures
References
-
- Sarafoglou K., Hoffmann G.F., Roth K.S. McGraw-Hill Company; New York: 2009. Pediatric Endocrinology and Inborn Errors of Metabolism; pp. 738–739. (& 744–745)
-
- Callahan J.W. Molecular basis of GM1 gangliosidosis and Morquio disease, type B. structure-function studies of lysosomal beta-galactosidase and the non-lysosomal beta-galactosidase-like protein. Biochim. Biophys. Acta. 1999;1455:85–103. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
