

Phosphatidylinositol 4,5-bisphosphate controls Rab7 and PLEKHM1 membrane cycling during autophagosome-lysosome fusion
- PMID: 31368593
- PMCID: PMC6463214
- DOI: 10.15252/embj.2018100312
Phosphatidylinositol 4,5-bisphosphate controls Rab7 and PLEKHM1 membrane cycling during autophagosome-lysosome fusion
Abstract
The small GTPase Rab7 is a key organizer of receptor sorting and lysosomal degradation by recruiting of a variety of effectors depending on its GDP/GTP-bound state. However, molecular mechanisms that trigger Rab7 inactivation remain elusive. Here we find that, among the endosomal pools, Rab7-positive compartments possess the highest level of PI4P, which is primarily produced by PI4K2A kinase. Acute conversion of this endosomal PI4P to PI(4,5)P2 causes Rab7 dissociation from late endosomes and releases a regulator of autophagosome-lysosome fusion, PLEKHM1, from the membrane. Rab7 effectors Vps35 and RILP are not affected by acute PI(4,5)P2 production. Deletion of PI4K2A greatly reduces PIP5Kγ-mediated PI(4,5)P2 production in Rab7-positive endosomes leading to impaired Rab7 inactivation and increased number of LC3-positive structures with defective autophagosome-lysosome fusion. These results reveal a late endosomal PI4P-PI(4,5)P2 -dependent regulatory loop that impacts autophagosome flux by affecting Rab7 cycling and PLEKHM1 association.
Keywords: Rab7; autophagy; lysosome; phosphatidylinositol 4-kinase; phosphoinositide.
Published 2019. This article is a U.S. Government work and is in the public domain in the USA.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

Cartoon depicting the principle of the endosomal PI4P BRET measurement. The Venus fluorescent protein is targeted to the respective endosomes using Rab proteins, while Renilla luciferase [termed Super luciferase (Sluc)] is fused to the P4M2x PI4P reporter. This reporter binds to PI4P the majority of which is in the PM. The PI4KA inhibitor, GSK‐A1 (A1), reduces PI4P in the PM allowing more of the reporter to find the endosomal PI4P that is produced by PI4Ks that are insensitive to GSK‐A1. Relocation of the P4M2x reporter tagged with Sluc to the PI4P‐rich endosomes will increase the energy transfer between the enzyme and Venus in the presence of the coelenterazine H substrate.
Representative confocal images show the distribution of the P4M2x reporter in HEK293‐AT1 cells before and after the addition of GSK‐A1 (30 nM). Note the signal disappearing from the PM and strongly highlighting the endosomes and Golgi. Scale bars are 20 μm.
Representative result from a BRET experiment detecting PI4P in various endosomes (Rab7, Rab5, and Rab11) following addition of GSK‐A1. The graph shows normalized BRET values where the averages of triplicate measurements of A1‐treated cells were divided by the averages of triplicates obtained in DMSO‐treated controls. Note that these signals originate from thousands of cells.
Similar BRET experiment showing the Rab7‐pool of PI4P both in control HEK293‐AT1 cells and in two clones of cells with PI4K2A deletion (#20 and #26). Means ± SEM are shown from three experiments performed in triplicates.
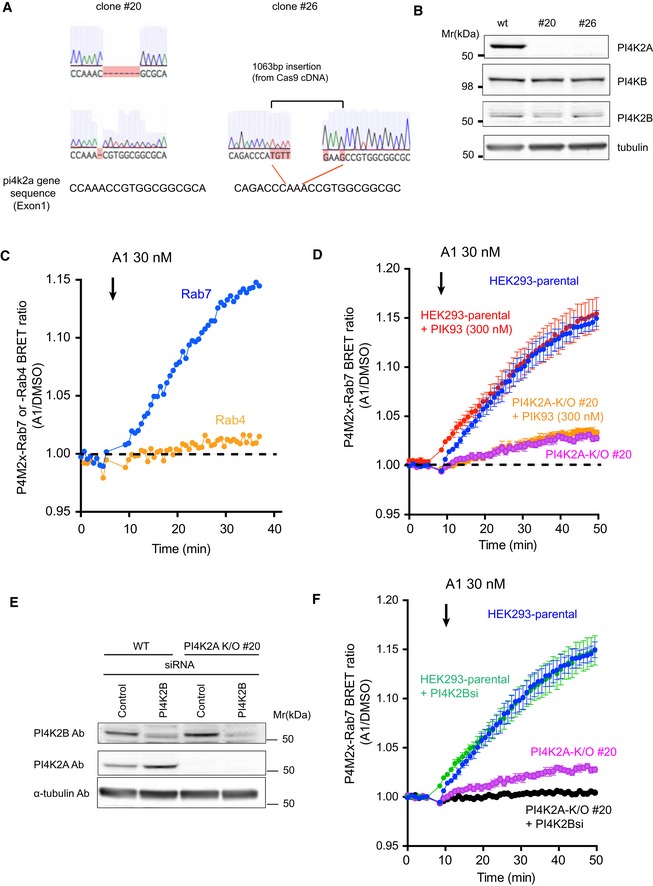
Sequence analysis of the targeted region in PI4K2A K/O #20 or #26 cells created with CRISPR/CAS9 genome editing system. The cartoons showing the altered sequences were captured from Benchling (
https://benchling.com/ ).The effect of PI4K2A K/O on the expression level of PI4KB and PI4K2B. Whole‐cell lysates prepared from wild‐type or the PI4K2A clones were subjected to Western blot analysis using anti‐PI4K2A, anti‐PI4KB, anti‐PI4K2B, and anti‐alpha‐tubulin antibodies.
Representative BRET measurement showing the relative amount of PI4P in Rab7‐ and Rab4‐positive endosomes. The BRET analysis was performed using the P4M2x‐Rab7 sensor as described in Fig 1C and D or by using P4M2x‐Rab4 sensor in which the Rab7 was replaced by the Rab4 sequence. A1 inhibitor was added to the cell following baseline measurement. The result shown is from an experiment performed in triplicates. The graph shows normalized BRET values where the averages of triplicate measurements of A1‐treated cells were divided by the averages of triplicates obtained in DMSO‐treated controls.
A BRET analysis was performed with the P4M2x‐Rab7 probe as described in Fig 1D. To inhibit PI4KB, 300 nM of PIK93 was added together with 30 nM of A1 following baseline measurements.
Wild‐type or PI4K2A K/O clone #20 cells were transfected with control siRNA or one targeting PI4K2B. Western blot analysis was performed with anti‐PI4K2B, anti‐PI4K2A, and anti‐alpha‐tubulin antibodies 3 days after the transfection.
A BRET analysis measuring late endosomal PI4P in cells after PI4K2B was knocked down in wild‐type or PI4K2A K/O cells. The values in (D) and (F) were measured in the same 96‐well plate but plotted separately for clarity (therefore, the blue curves are the same in the two panels). The BRET ratios were expressed relative to those of DMSO‐treated control cells, and means ± SEM are shown from three experiments performed in triplicates.
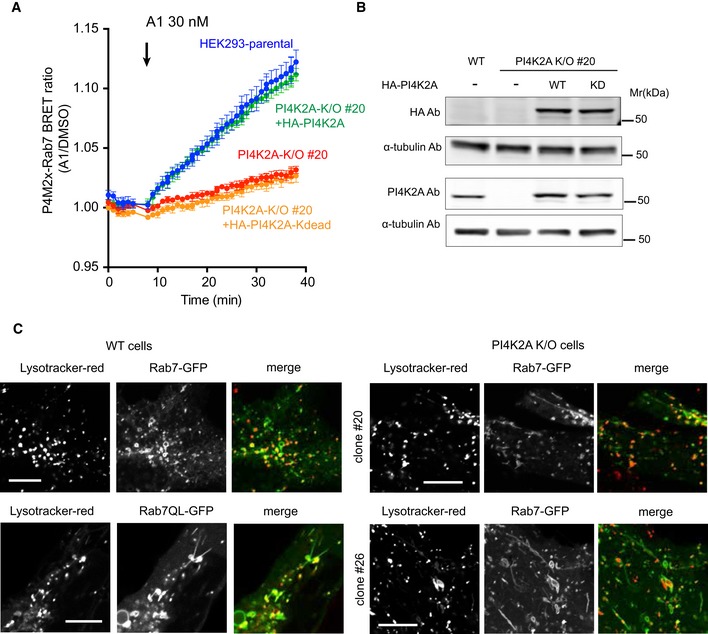
BRET measurement of PI4P in Rab7 compartment in parental and PI4K2A K/O cells after expression of HA‐tagged PI4K2A or its kinase‐dead mutant. Means ± SEM are shown from three experiments performed in triplicates.
Western blot analysis shows that the level of PI4K2A expression was closely matched with that of wild‐type cells.
Distribution of GFP‐Rab7 in parental (left panels) and two clones of PI4K2A K/O cells. Representative pictures show tubulation in the K/O cells reminiscent of those seen in wild‐type cells expressing the GTP‐locked Rab7Q67L mutant. Scale bars are 10 μm.
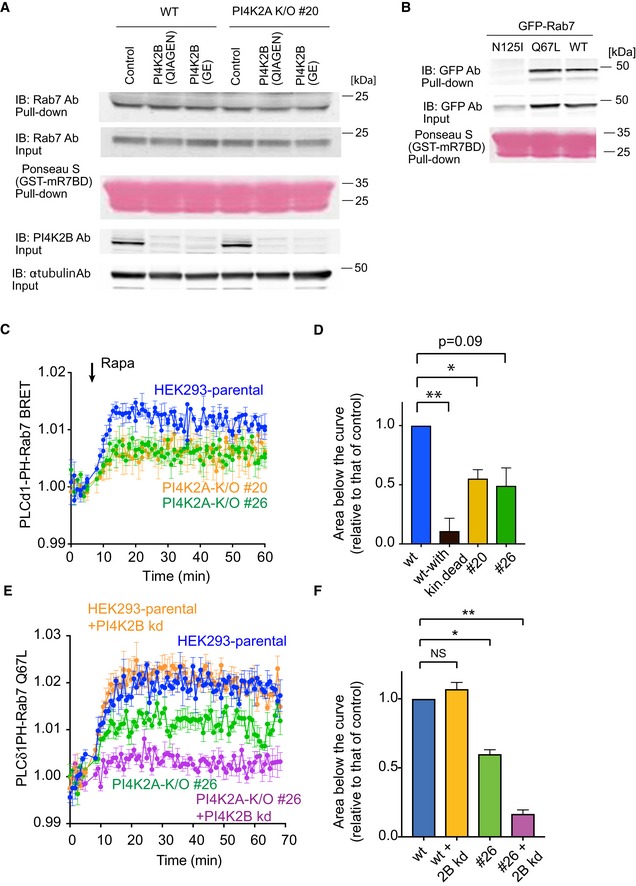
Pull‐down assays were performed as described in Materials and Methods using lysates prepared from wild‐type or PI4K2A K/O cells #20 in which PI4K2B was knocked down by two different siRNAs (obtained from QIAGEN or GE). Whole‐cell lysates were collected 3 days after the transfection and active Rab7 captured by the Rab7‐GTP interacting motif of RILP. Western blot analysis of the captured material was performed with anti‐Rab7, anti‐PI4K2B, and anti‐alpha‐tubulin antibodies.
Pull‐down assays showing the amount of overexpressed Rab7 wild‐type, N125I, Q67L mutant captured by the Rab7‐GTP binding domain.
Increased production of PI(4,5)P2 in the Rab7 compartment by a recruited PIP5Kγ enzyme measured by BRET analysis in control and PI4K2A K/O cells. This panel originates from the same experiments shown in Fig 3C plotted separately for clarity (therefore, blue control curves are the same). For experimental details, see legend to Fig 3C. Means ± SEM from three separate experiments each performed in triplicates.
This bar diagram shows the areas below the curves calculated from the time of rapamycin addition for each of four separate experiments similar to those shown in panel C (means ± SEM, n = 4). One‐way ANOVA with Dunnett's multiple comparisons was used for statistical analysis (*P = 0.0204; **P = 0.0081).
Knockdown (kd) of PI4K2B further reduces the amount of PI(4,5)P2 generated by a recruited PIP5Kγ in the Rab7 compartment. This experiment was performed similarly to that shown in Fig 3E using the Rab7‐QL mutant in the BRET construct. Means ± SEM from three separate experiments each performed in triplicates.
Quantitation and statistical analysis of data shown in panel (E) performed as described for panel (D) (*P = 0.0118; **P = 0.0023).

Cartoon depicting the experimental design. A PIP5Kγ construct fused to FKBP12 is expressed together with an FRB module fused to Rab7 and the GFP‐tagged PLCδ1PH reporter for PI(4,5)P2. Addition of rapamycin causes recruitment of PIP5Kγ to the endosomes where it makes more PI(4,5)P2 and attracts the PI(4,5)P2 reporter.
Representative confocal pictures showing these changes in live cells. Cells were transfected with the indicated constructs and imaged before and after addition of rapamycin (100 nM) for 10 min. Note that the Rab7 recruiter itself is dissociating from the membranes after PI(4,5)P2 generation. Left panels show some cells enlarged. Scale bars: 20 μm.
Quantification of the changes in BRET experiments. Here the BRET sensor contained the PLCδ1PH‐fused to Sluc and the Venus targeted with Rab7 expressed from a single vector and it was co‐transfected with the Rab7‐targeted FRB (tagged with iRFP) and the FKBP12‐fused PIP5Kγ in which the CFP was mutated to eliminate its fluorescence (CFP*). Recruitment of the PIP5Kγ but not its kinase‐dead version caused an increase in the BRET signal indicating the increased PI(4,5)P2 in this compartment (means ± SEM, from three separate experiments performed in triplicates).
PI(4,5)P2 increases were larger when the Venus part of the BRET sensor was targeted with the GTP‐locked form of Rab7 (Q67L) and was negligible with the GDP‐locked form of Rab7 (N125I) in the BRET construct. Also note that the Rab7 wild‐type‐based BRET signal slowly returned toward the green trace consistent with Rab7 falling off from the membrane both in the BRET and the recruiting constructs (means ± SEM, from three separate experiments performed in triplicates).
The response of PI4K2A K/O cells was significantly reduced in these latter experiments. Means ± SEM are shown from three experiments performed in triplicates.
Areas below the curves calculated from the time of rapamycin addition for each of the three separate experiments shown in panel E (means ± SEM, n = 3). One‐way ANOVA with Dunnett's multiple comparisons was used for statistical analysis (*P = 0.0478 and 0.0287 for #20 and #26 clones, respectively).
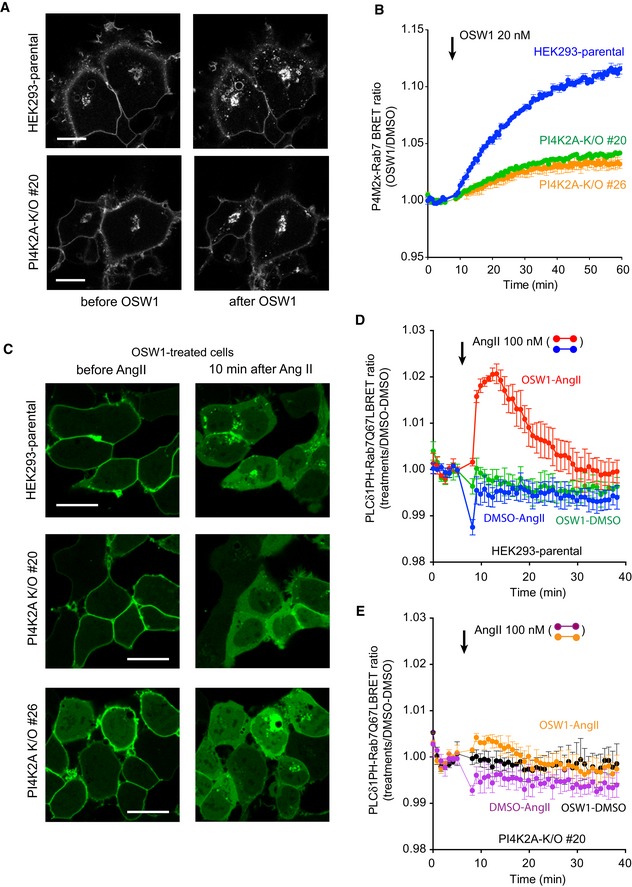
Representative confocal images of HEK293‐AT1 cells, which show that treatment with OSW1 (20 nM), a drug that inhibits the cholesterol‐PI4P transport protein, OSBP, causes accumulation of PI4P in the Golgi and in the endosomes (upper panels). Note that the accumulation is much reduced in the endosome but less so in the Golgi in the PI4K2A K/O cells. Scale bars: 20 μm.
Quantification of these changes by BRET analysis. The PI4P‐Rab7 BRET sensor construct was transfected into the respective cell lines and the cells treated with OSW1 (20 nM) for the indicated times. The BRET ratios were expressed relative to those of DMSO‐treated cells.
Detection of endosomal PI(4,5)P2 in cells pretreated with OSW1 for 1 h in serum‐free medium using confocal microscopy and the PLCδ1PH‐GFP PI(4,5)P2 reporter. Angiotensin II (AngII, 100 nM; a Gq and PLC‐activating agonist) was used to liberate the PLCδ1PH‐GFP PI(4,5)P2 reporter from the PM, which was necessary to detect the endosomal PI(4,5)P2. The signal is transient as the AngII receptors show desensitization. Note the reduced signal in the two PI4K2A K/O clones. Scale bars: 20 μm.
Quantification of the same changes by BRET analysis in wild‐type cells using the PI(4,5)P2 sensor with Rab7‐Q67L‐targeting. Note the transient signal increase after AngII stimulation only in cells treated with OSW1.
This increase is barely detectable in the PI4K2A K/O cells.
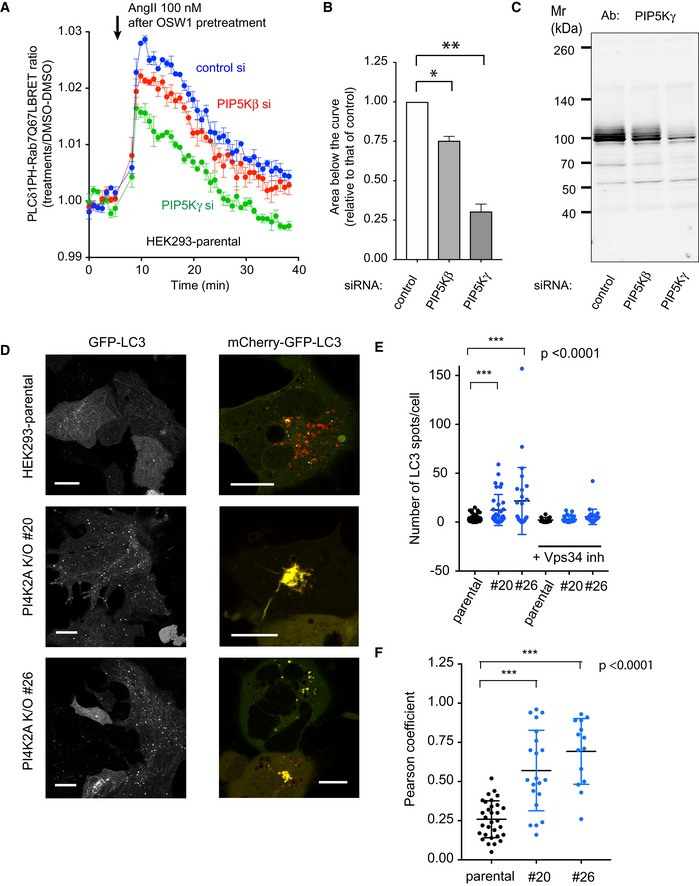
BRET experiment in parental HEK293‐AT1 cells in OSW1‐pretreated cells after RNAi‐mediated knockdown of PIP5Kβ or PIP5Kγ. The BRET experiment was performed the same way as described in the legend to Fig 4D (means ± SEM from three experiments, each performed in triplicates). Note the reduced response in the PIP5Kγ knockdown cells and the slight reduction in the case of PIP5Kβ knockdown.
Areas below the curves calculated from the time of rapamycin addition for each of the three separate experiments shown in panel A (means ± SEM, n = 3). One‐way ANOVA with Dunnett's multiple comparisons was used for statistical analysis (*P = 0.0241; **P = 0.0088).
These reductions are proportional to the reduction in PIP5Kγ levels as assessed by Western blot analysis of cell lysates obtained from cells treated with siRNAs at the same time.
Increased appearance of GFP‐LC3‐positive vesicles in PI4K2A K/O cells and reduced acidification of the LC3‐positive vesicles using the GFP‐mCherry‐LC3 reporter. Cells were transfected with the indicated constructs for 1 day and observed by confocal microscopy. Scale bars: 20 μm.
Quantification of the LC3‐positive vesicles (n = 67, 34, 26, 13, 30, 27 for the various groups, respectively, from left to right). *** designates P < 0.0001 using unpaired t‐test. The same data set was also analyzed with the non‐parametric Mann–Whitney U‐test showing a P = 0.0004 and 0.0012 for #20 and #26 clones relative to control, respectively.
Co‐localization analysis using the Pearson coefficient (n = 31, 21, and 14 cells analyzed for the different groups from left to right). *** designates P < 0.0001 using unpaired t‐test. The same data set was also analyzed with the non‐parametric Mann–Whitney U‐test showing a P value < 0.0001 for both groups.

- A, C
Western blot analysis with two different PIP5Kβ antibodies (AB1‐ or AB2‐PIP5Kβ) performed with whole‐cell lysates prepared from wild‐type cells or from cells expressing myc‐ or mRFP‐tagged PIP5Kβ. Note that both antibodies detect the expressed proteins but failed to detect the endogenous PIP5Kβ.
- B
siRNA against PIP5Kβ is effective as assessed by Western blotting using a GFP‐humanPIP5Kβ enzyme. The amounts of transfected PIP5Kβ plasmid DNA are indicated.
- D
Western blot analysis showing the lack of effect of PIP5Kβ or PIPK5γ siRNA on the amount of PIP5Kα.
- E
BRET analysis showing the level of changes in plasma membrane PI(4,5)P2 during stimulation by AngII in cells depleted in PIP5Kβ or PIPK5γ. AngII was added to the cells for monitoring the rate of PI(4,5)P2 breakdown and subsequent re‐synthesis. The BRET ratios were expressed relative to those of DMSO‐treated cells, and means ± SEM are shown from three experiments performed in triplicates.

- A, B
Distribution of mCherry‐GFP‐LC3 in HEK293‐AT1 cells (without starvation) after treatment with control siRNA (A) or siRNA for PIP5Kγ (PIP5K1C). Scale bars: 20 μm. Note the massive tubulation of the LC3 compartment in some of the knock‐down cells (B, lower images, scale bar: 10 μm).
- C
Comparison of Pearson coefficients from cells treated with control or PIP5Kγ RNAi. For statistical analysis, the unpaired t‐test was used (n = 50 and 45 cells for control a PIP5Kγ knockdown groups, respectively; ***P < 0.0001).
- D
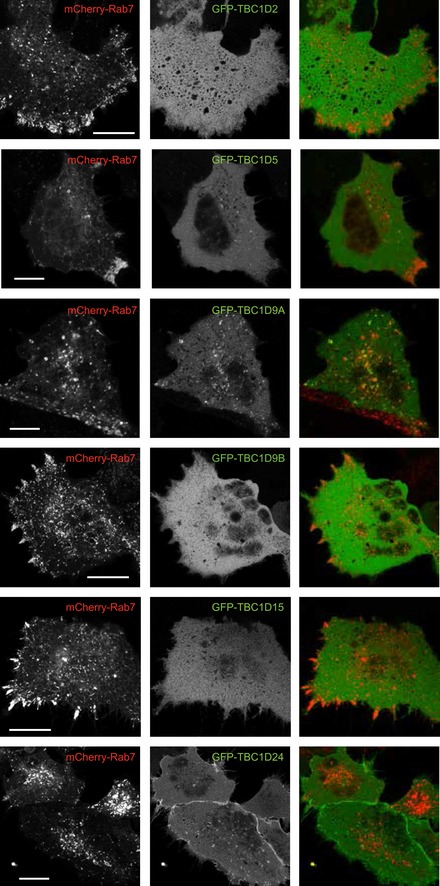
Representative confocal images of live HEK293‐AT1 cells expressing GFP‐Rab7 treated with control or PIP5Kγ RNAi. Note the tubulation in the PIP5Kγ RNAi‐treated cells. Scale bars: 10 μm.

HEK293‐AT1 cells were transfected with PLEKHM1‐GFP‐Rab7 along with CFP‐FKBP‐PIP5Kγ and iRFP‐FRB‐Rab7 constructs for 1 day and examined live by confocal microscopy. Representative cells show reduced PLEKHM1 localization when PIP5Kγ was recruited to the Rab7 endosomes (lower row images).
This response is reduced in PI4K2A K/O cells.
Quantification of these changes by BRET analysis where the full‐length PLEKHM1 was fused to Sluc and Venus was fused to Rab7 in the BRET construct. For PIP5Kγ recruitment, a mutant dark CFP(W66A)‐FKBP‐PIP5Kγ was used with an iRFP‐FRB‐Rab7. BRET values were normalized to those of DMSO‐treated cells. Comparisons were made for parental (blue) and PI4K2A K/O (green and orange) cells.
Similar BRET analysis where the PLEKHM1 was replaced by either RILP or Vps35 in the BRET construct.

- A
BRET analysis was performed as described for Fig 7C except that the RH domain of PLEKHM1 instead of the full‐length PLEKHM1 protein was used in the BRET construct. Recruitment of PIP5Kγ to the Rab7 endosomes caused dissociation of the RH domain from late endosomes. This response was larger in wild‐type cells than in PI4K2A K/O #20 cells. Means ± SEM from three separate experiments each performed in triplicates.
- B
BRET analysis showing that when Rab7Q67L mutant was used in the BRET construct it failed to release either the full‐length PLEKHM1 or its RH domain from late endosomes when PI(4,5)P2 was acutely produced. CFP(W66A)‐FKBP‐PIP5Kγ and iRFP‐FRB‐Rab7 was used as the recruitment constructs and Sluc‐fused full‐length PLEKHM1 or its RH domain together with Venus‐tagged Rab7wt or Q67L mutant as the BRET sensor. Means ± SEM from three separate experiments each performed in triplicates.
- C, D
Live‐cell confocal imaging showing the distribution of GFP‐RILP or GFP‐Vps35 upon acute production of PI(4,5)P2 in the Rab7 compartment. HEK293‐AT1 cells were transfected with CFP‐FKBP‐PIP5Kγ, iRFP‐FRB‐Rab7, and GFP‐RILP(C) or GFP‐Vps35(D). Next day, the images were captured by confocal microscopy before and after 100 nM rapamycin treatment. Scale bars: 20 μm.
References
-
- Balla A, Tuymetova G, Barshishat M, Geiszt M, Balla T (2002) Characterization of type II phosphatidylinositol 4‐kinase isoforms reveals association of the enzymes with endosomal vesicular compartments. J Biol Chem 277: 20041–22050 - PubMed
-
- Bo T, Yan F, Guo J, Lin X, Zhang H, Guan Q, Wang H, Fang L, Gao L, Zhao J, Xu C (2016) Characterization of a relatively malignant form of osteopetrosis caused by a novel mutation in the PLEKHM1 gene. J Bone Miner Res 31: 1979–1987 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
