

Metabolic Plasticity of Acute Myeloid Leukemia
- PMID: 31370337
- PMCID: PMC6721808
- DOI: 10.3390/cells8080805
Metabolic Plasticity of Acute Myeloid Leukemia
Abstract
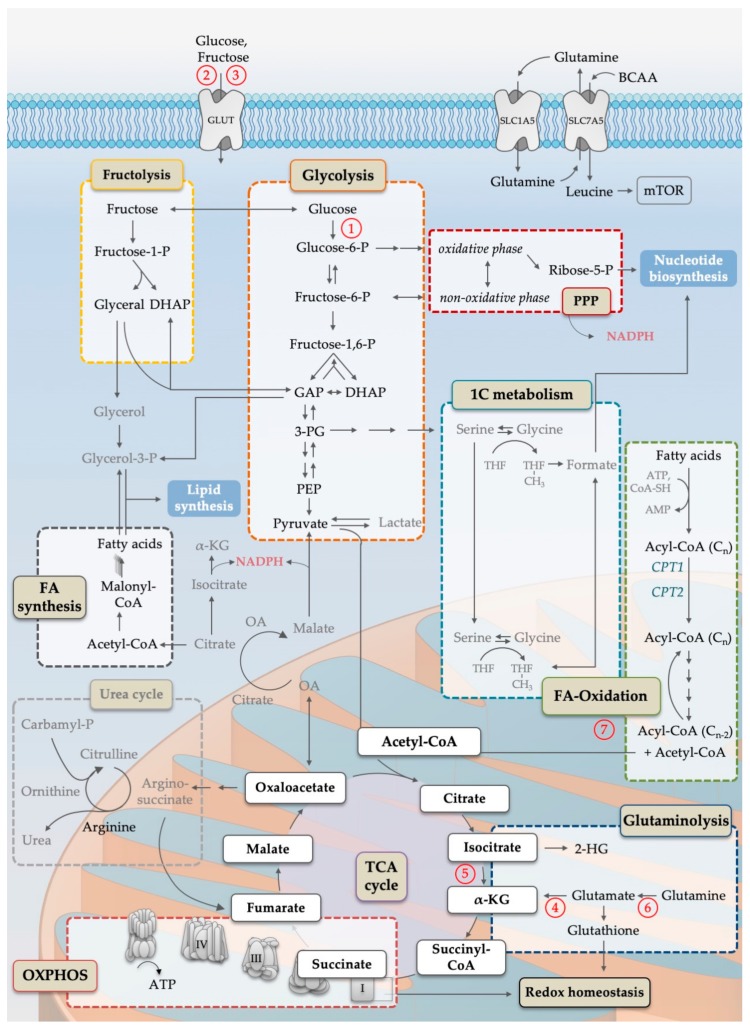
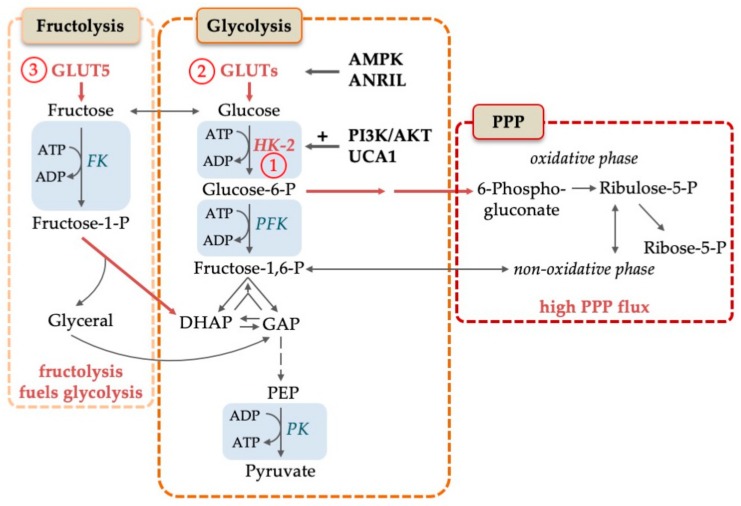
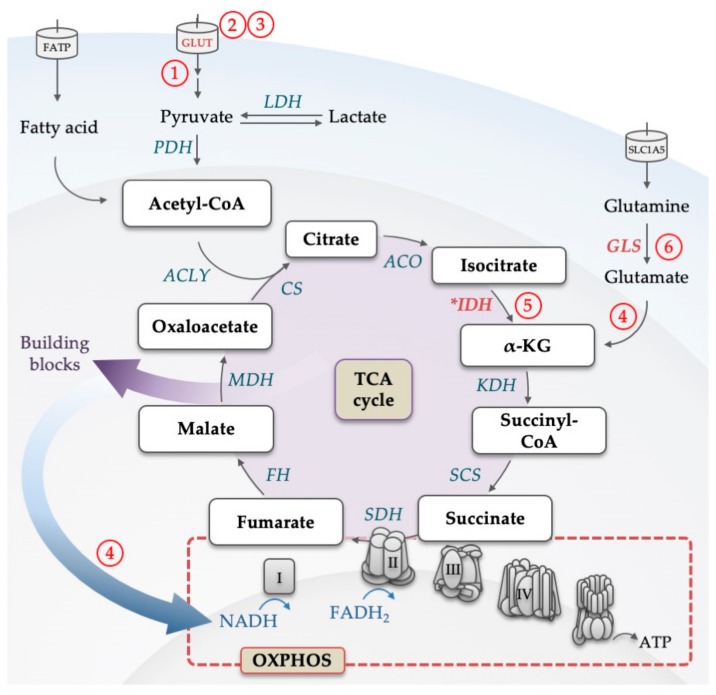
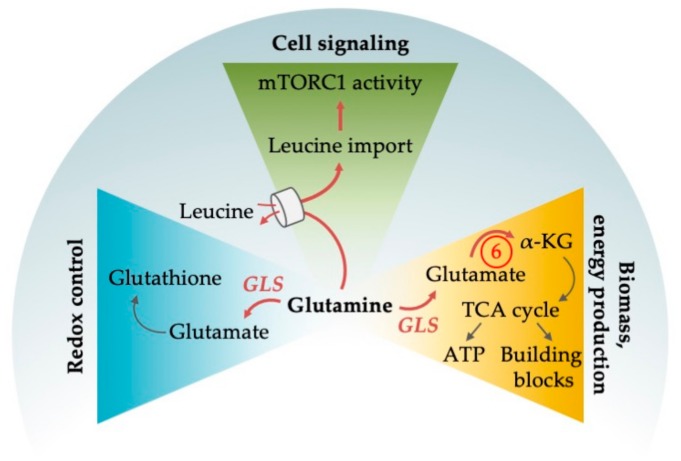
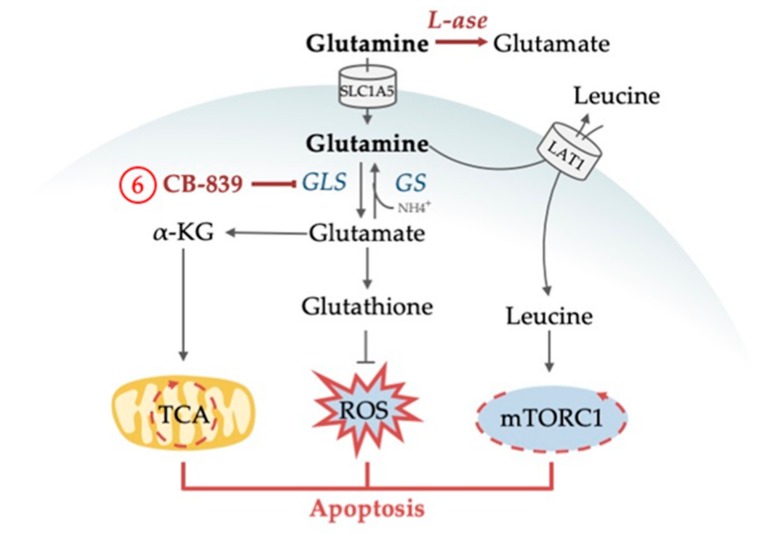
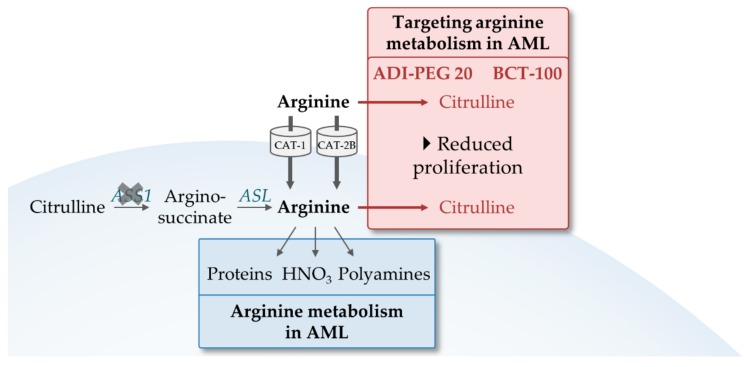
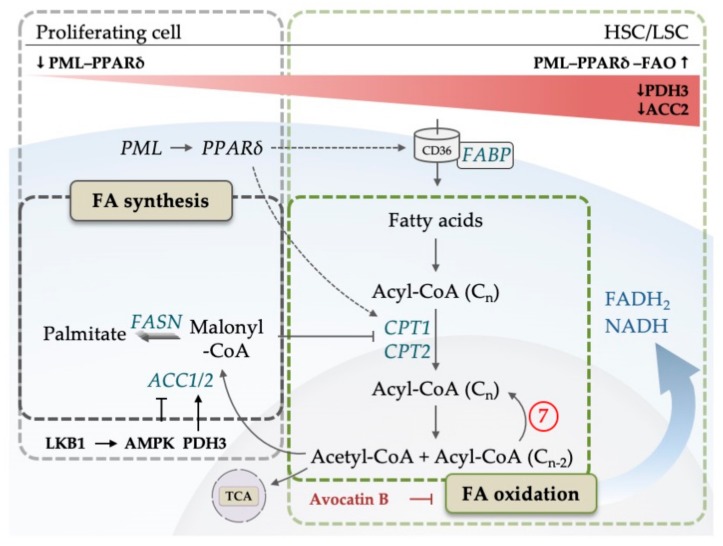
Acute myeloid leukemia (AML) is one of the most common and life-threatening leukemias. A highly diverse and flexible metabolism contributes to the aggressiveness of the disease that is still difficult to treat. By using different sources of nutrients for energy and biomass supply, AML cells gain metabolic plasticity and rapidly outcompete normal hematopoietic cells. This review aims to decipher the diverse metabolic strategies and the underlying oncogenic and environmental changes that sustain continuous growth, mediate redox homeostasis and induce drug resistance in AML. We revisit Warburg's hypothesis and illustrate the role of glucose as a provider of cellular building blocks rather than as a supplier of the tricarboxylic acid (TCA) cycle for energy production. We discuss how the diversity of fuels for the TCA cycle, including glutamine and fatty acids, contributes to the metabolic plasticity of the disease and highlight the roles of amino acids and lipids in AML metabolism. Furthermore, we point out the potential of the different metabolic effectors to be used as novel therapeutic targets.
Keywords: TCA cycle; acute myeloid leukemia; aerobic glycolysis; amino acids; drug resistance; fatty acids; leukemic stem cells; metabolic plasticity; oxidative phosphorylation; redox homeostasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Arber D.A. Acute Myeloid Leukemia. In: Longo D.L., editor. Hematopathology: A Volume in the Series: Foundations in Diagnostic Pathology. Volume 12. Massachusetts Medical Society; Waltham, MA, USA: 2017. pp. 429–466.e5.
-
- Cancer Genome Atlas Research, Network. Ley T.J., Miller C., Ding L., Raphael B.J., Mungall A.J., Robertson A.G., Hoadley K., Triche T.J., Laird P.W., et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013;368:2059–2074. doi: 10.1056/nejmoa1301689. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
