

A hybrid, bottom-up, structurally accurate, Go¯-like coarse-grained protein model
- PMID: 31370551
- PMCID: PMC6663515
- DOI: 10.1063/1.5108761
A hybrid, bottom-up, structurally accurate, Go¯-like coarse-grained protein model
Abstract
Coarse-grained (CG) protein models in the structural biology literature have improved over the years from being simple tools to understand general folding and aggregation driving forces to capturing detailed structures achieved by actual folding sequences. Here, we ask whether such models can be developed systematically from recent advances in bottom-up coarse-graining methods without relying on bioinformatic data (e.g., protein data bank statistics). We use relative entropy coarse-graining to develop a hybrid CG but Go¯-like CG peptide model, hypothesizing that the landscape of proteinlike folds is encoded by the backbone interactions, while the sidechain interactions define which of these structures globally minimizes the free energy in a unique native fold. To construct a model capable of capturing varied secondary structures, we use a new extended ensemble relative entropy method to coarse-grain based on multiple reference atomistic simulations of short polypeptides with varied α and β character. Subsequently, we assess the CG model as a putative protein backbone forcefield by combining it with sidechain interactions based on native contacts but not incorporating native distances explicitly, unlike standard Go¯ models. We test the model's ability to fold a range of proteins and find that it achieves high accuracy (∼2 Å root mean square deviation resolution for both short sequences and large globular proteins), suggesting the strong role that backbone conformational preferences play in defining the fold landscape. This model can be systematically extended to non-natural amino acids and nonprotein polymers and sets the stage for extensions to non-Go¯ models with sequence-specific sidechain interactions.
Figures









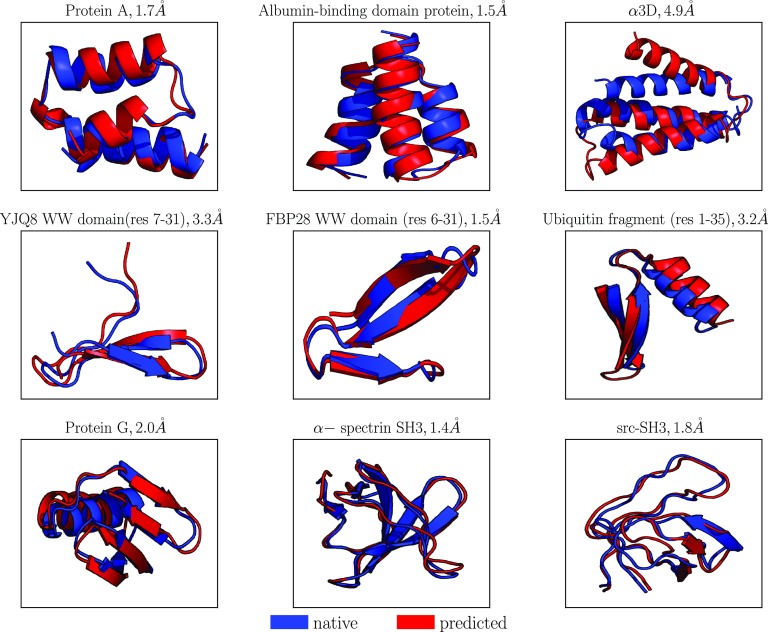

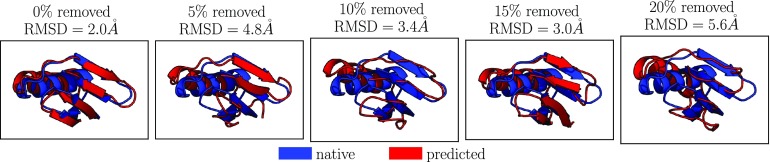
References
-
- Brito J. A. and Archer M., “X-ray crystallography,” in Practical Approaches to Biological Inorganic Chemistry, edited by Crichton R. R. and Louro R. O. (Elsevier, Oxford, 2013), Chap. 9, pp. 217–255.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources