

Production of seedable Amyloid-β peptides in model of prion diseases upon PrPSc-induced PDK1 overactivation
- PMID: 31371707
- PMCID: PMC6672003
- DOI: 10.1038/s41467-019-11333-3
Production of seedable Amyloid-β peptides in model of prion diseases upon PrPSc-induced PDK1 overactivation
Abstract
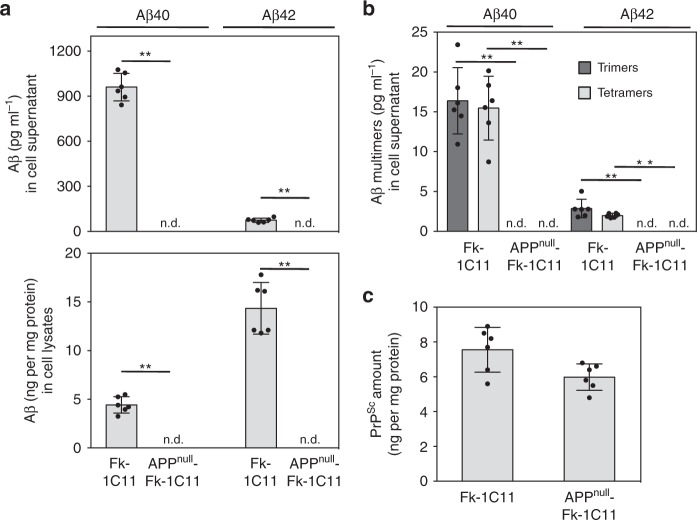
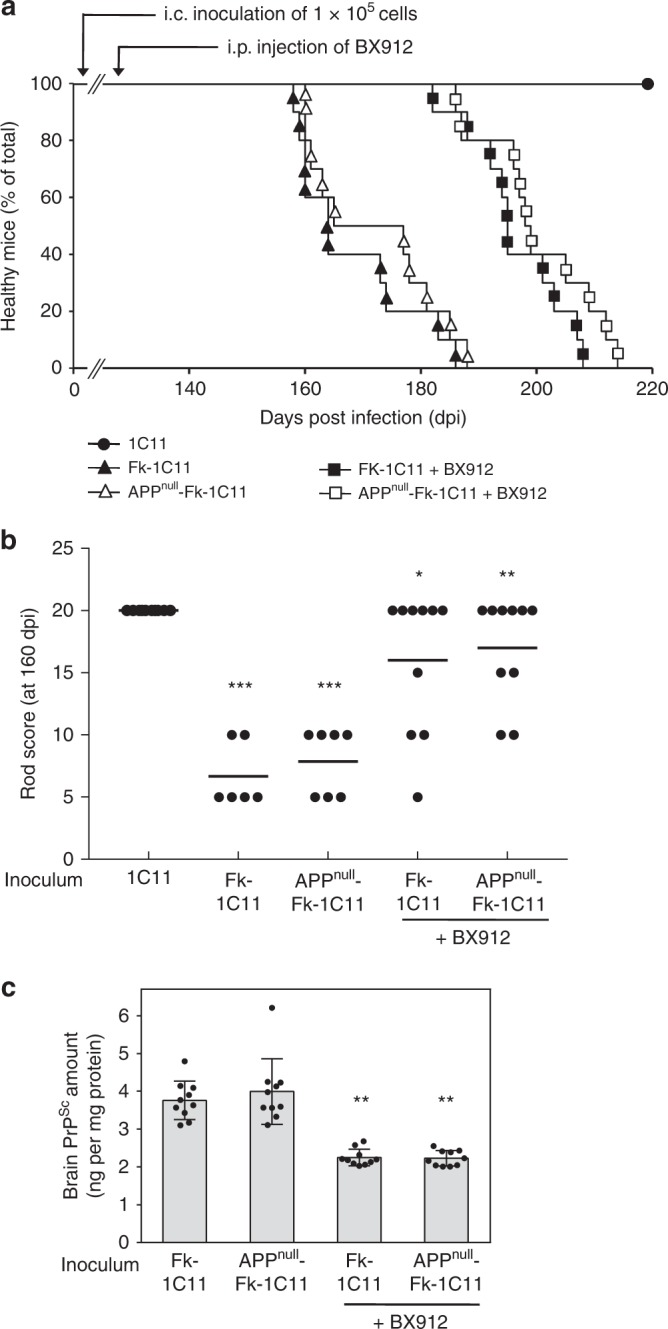
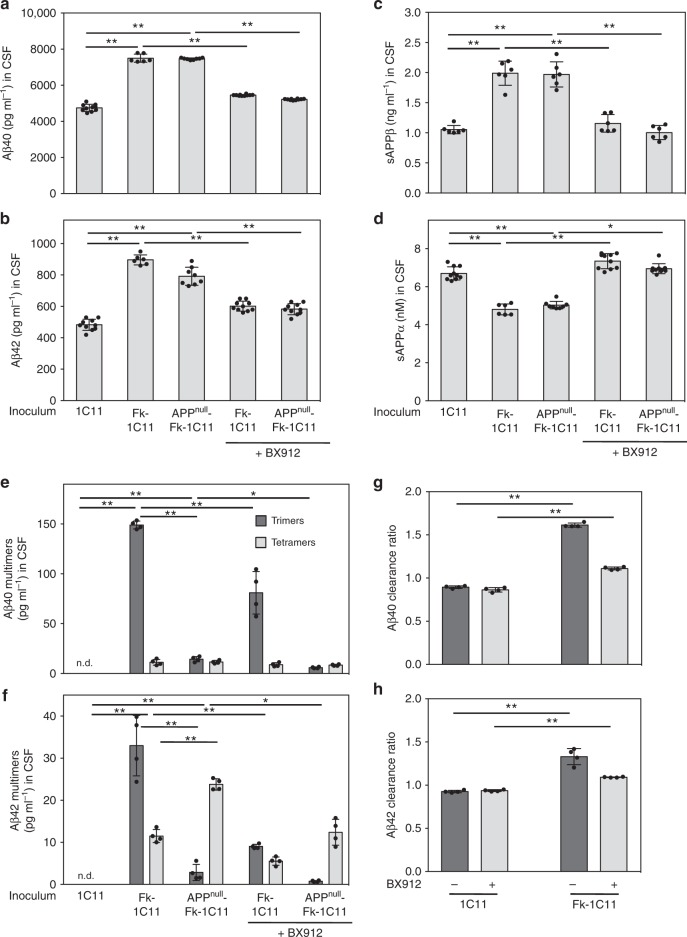
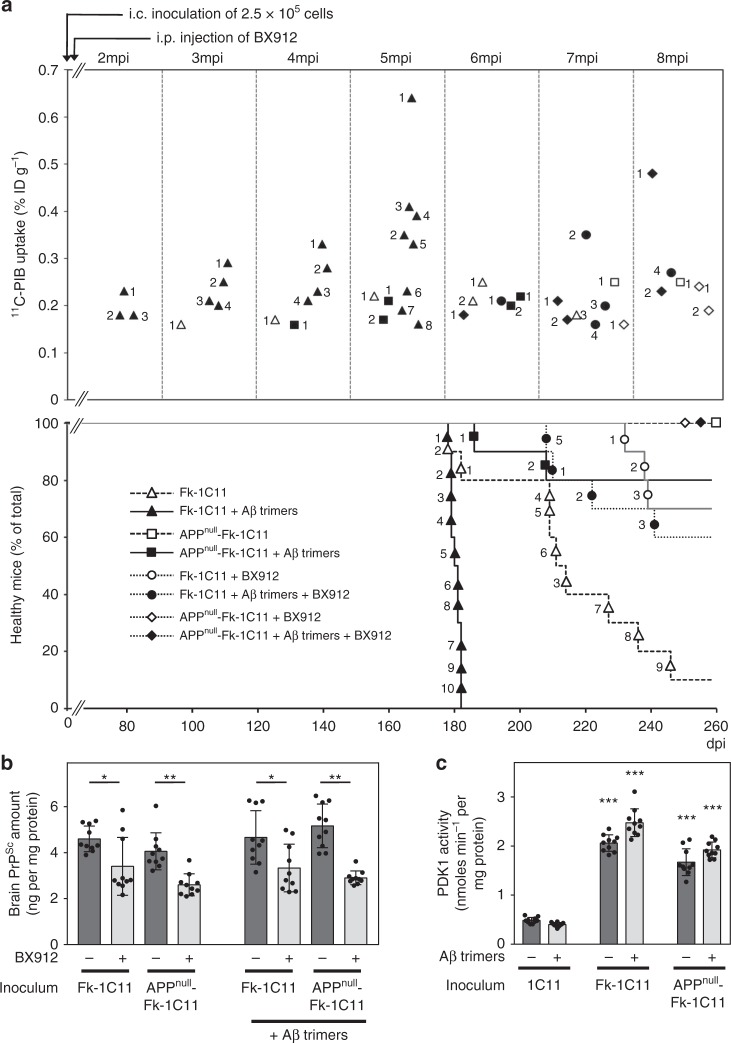
The presence of amyloid beta (Aβ) plaques in the brain of some individuals with Creutzfeldt-Jakob or Gertsmann-Straussler-Scheinker diseases suggests that pathogenic prions (PrPSc) would have stimulated the production and deposition of Aβ peptides. We here show in prion-infected neurons and mice that deregulation of the PDK1-TACE α-secretase pathway reduces the Amyloid Precursor Protein (APP) α-cleavage in favor of APP β-processing, leading to Aβ40/42 accumulation. Aβ predominates as monomers, but is also found as trimers and tetramers. Prion-induced Aβ peptides do not affect prion replication and infectivity, but display seedable properties as they can deposit in the mouse brain only when seeds of Aβ trimers are co-transmitted with PrPSc. Importantly, brain Aβ deposition accelerates death of prion-infected mice. Our data stress that PrPSc, through deregulation of the PDK1-TACE-APP pathway, provokes the accumulation of Aβ, a prerequisite for the onset of an Aβ seeds-induced Aβ pathology within a prion-infectious context.
Conflict of interest statement
J.M.L has non-financial competing interests with Hoffmann La Roche Ltd laboratories. He acts as an expert witness for Hoffmann La Roche Ltd laboratories. This does not alter our adherence to all Nature Communications policies on sharing data and materials. The remaining authors declare no competing interests.
Figures


Similar articles
-
PDK1 decreases TACE-mediated α-secretase activity and promotes disease progression in prion and Alzheimer's diseases.Nat Med. 2013 Sep;19(9):1124-31. doi: 10.1038/nm.3302. Epub 2013 Aug 18. Nat Med. 2013. PMID: 23955714
-
Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein.Proc Natl Acad Sci U S A. 2007 Jun 26;104(26):11062-7. doi: 10.1073/pnas.0609621104. Epub 2007 Jun 15. Proc Natl Acad Sci U S A. 2007. PMID: 17573534 Free PMC article.
-
Prion infection of mice transgenic for human APPSwe: increased accumulation of cortical formic acid extractable Abeta(1-42) and rapid scrapie disease development.Int J Dev Neurosci. 2008 Nov;26(7):821-4. doi: 10.1016/j.ijdevneu.2008.07.001. Epub 2008 Jul 9. Int J Dev Neurosci. 2008. PMID: 18662767
-
Amyloid associated proteins in Alzheimer's and prion disease.Curr Drug Targets CNS Neurol Disord. 2005 Jun;4(3):235-48. doi: 10.2174/1568007054038184. Curr Drug Targets CNS Neurol Disord. 2005. PMID: 15975027 Review.
-
Roles of proteolysis and lipid rafts in the processing of the amyloid precursor protein and prion protein.Biochem Soc Trans. 2005 Apr;33(Pt 2):335-8. doi: 10.1042/BST0330335. Biochem Soc Trans. 2005. PMID: 15787600 Review.
Cited by
-
Post-translational modifications in prion diseases.Front Mol Neurosci. 2024 Jul 1;17:1405415. doi: 10.3389/fnmol.2024.1405415. eCollection 2024. Front Mol Neurosci. 2024. PMID: 39011540 Free PMC article. Review.
-
Loss of prion protein control of glucose metabolism promotes neurodegeneration in model of prion diseases.PLoS Pathog. 2021 Oct 5;17(10):e1009991. doi: 10.1371/journal.ppat.1009991. eCollection 2021 Oct. PLoS Pathog. 2021. PMID: 34610054 Free PMC article.
-
Titanium dioxide and carbon black nanoparticles disrupt neuronal homeostasis via excessive activation of cellular prion protein signaling.Part Fibre Toxicol. 2022 Jul 15;19(1):48. doi: 10.1186/s12989-022-00490-x. Part Fibre Toxicol. 2022. PMID: 35840975 Free PMC article.
-
ADAM Sheddase Activity Promotes the Detachment of Small Extracellular Vesicles From the Plasma Membrane.J Extracell Vesicles. 2025 Jul;14(7):e70114. doi: 10.1002/jev2.70114. J Extracell Vesicles. 2025. PMID: 40673783 Free PMC article.
-
The Cellular Prion Protein-ROCK Connection: Contribution to Neuronal Homeostasis and Neurodegenerative Diseases.Front Cell Neurosci. 2021 Apr 12;15:660683. doi: 10.3389/fncel.2021.660683. eCollection 2021. Front Cell Neurosci. 2021. PMID: 33912016 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
