

The role of GRHL2 and epigenetic remodeling in epithelial-mesenchymal plasticity in ovarian cancer cells
- PMID: 31372511
- PMCID: PMC6656769
- DOI: 10.1038/s42003-019-0506-3
The role of GRHL2 and epigenetic remodeling in epithelial-mesenchymal plasticity in ovarian cancer cells
Abstract
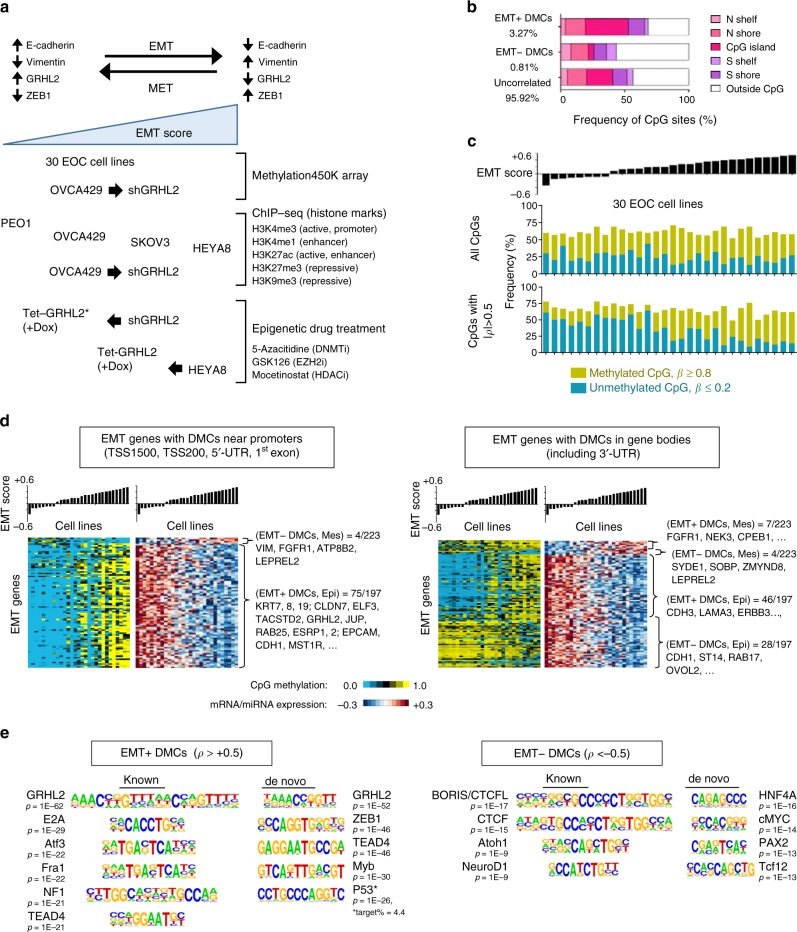
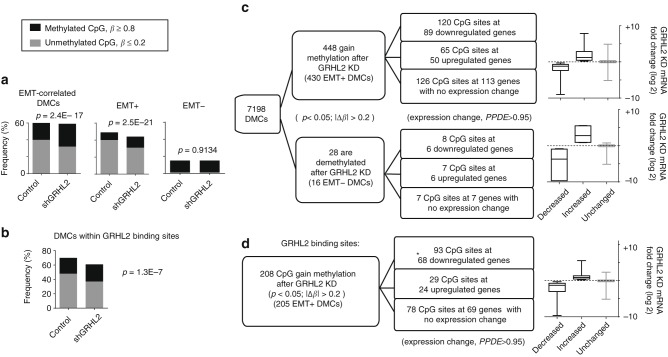
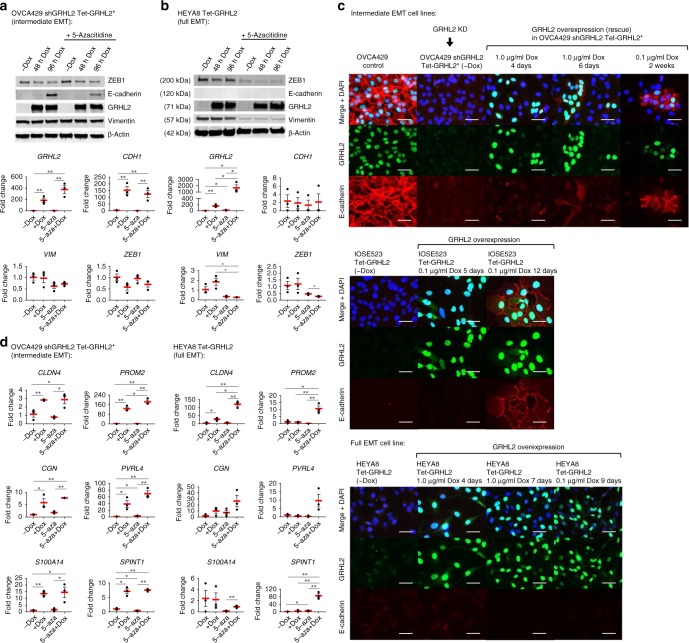
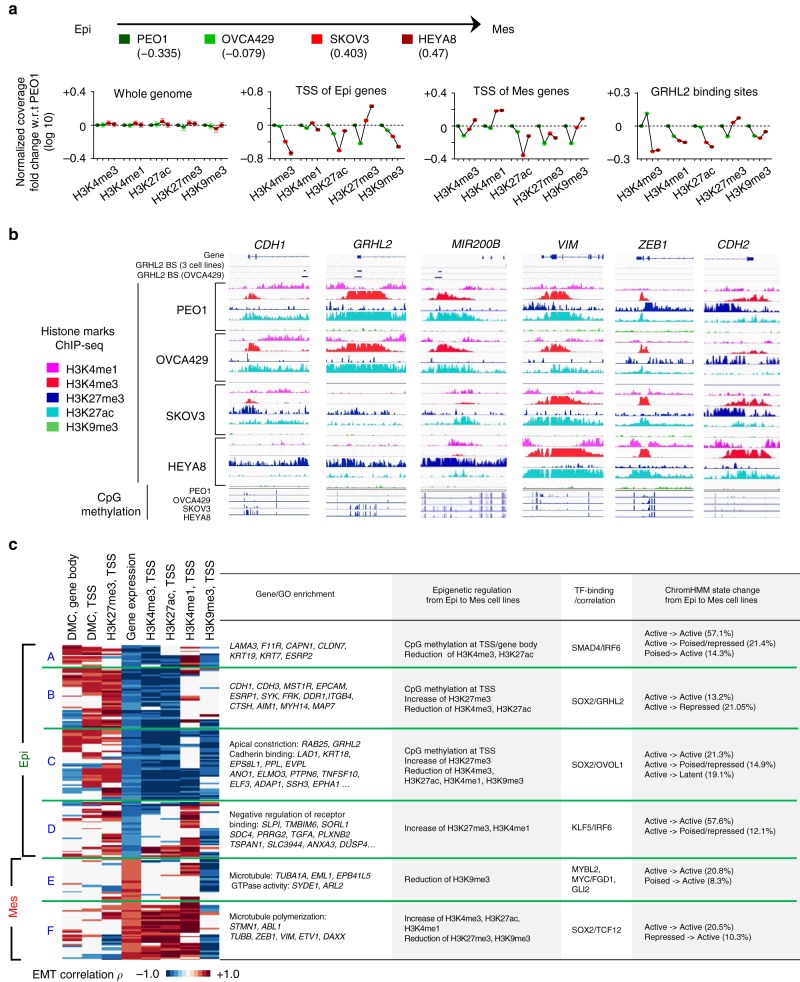
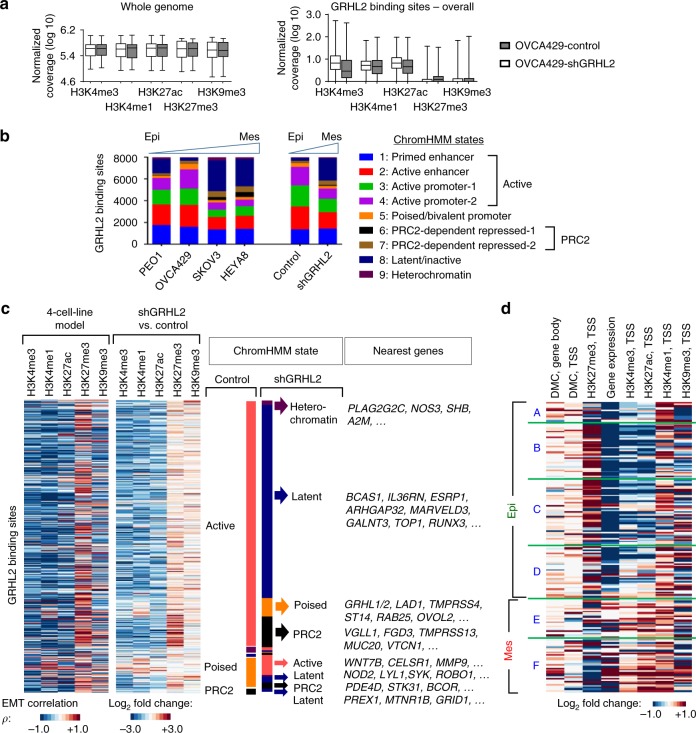
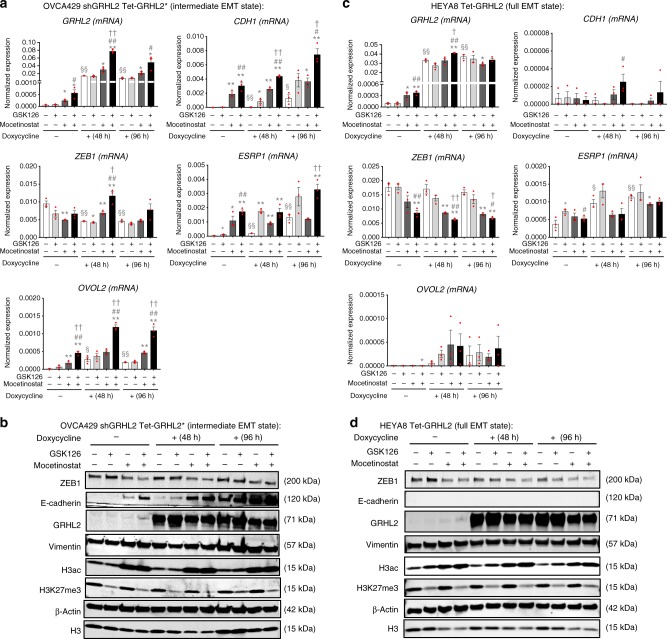
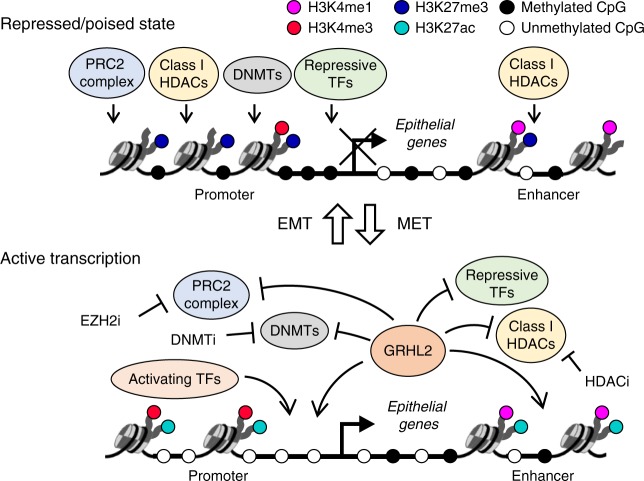
Cancer cells exhibit phenotypic plasticity during epithelial-mesenchymal transition (EMT) and mesenchymal-epithelial transition (MET) involving intermediate states. To study genome-wide epigenetic remodeling associated with EMT plasticity, we integrate the analyses of DNA methylation, ChIP-sequencing of five histone marks (H3K4me1, H3K4me3, H3K27Ac, H3K27me3 and H3K9me3) and transcriptome profiling performed on ovarian cancer cells with different epithelial/mesenchymal states and on a knockdown model of EMT suppressor Grainyhead-like 2 (GRHL2). We have identified differentially methylated CpG sites associated with EMT, found at promoters of epithelial genes and GRHL2 binding sites. GRHL2 knockdown results in CpG methylation gain and nucleosomal remodeling (reduction in permissive marks H3K4me3 and H3K27ac; elevated repressive mark H3K27me3), resembling the changes observed across progressive EMT states. Epigenetic-modifying agents such as 5-azacitidine, GSK126 and mocetinostat further reveal cell state-dependent plasticity upon GRHL2 overexpression. Overall, we demonstrate that epithelial genes are subject to epigenetic control during intermediate phases of EMT/MET involving GRHL2.
Keywords: Differentiation; Epigenetics; Ovarian cancer.
Conflict of interest statement
Competing interestsThe authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
