

Extensive transcriptional responses are co-ordinated by microRNAs as revealed by Exon-Intron Split Analysis (EISA)
- PMID: 31372646
- PMCID: PMC6895270
- DOI: 10.1093/nar/gkz664
Extensive transcriptional responses are co-ordinated by microRNAs as revealed by Exon-Intron Split Analysis (EISA)
Abstract
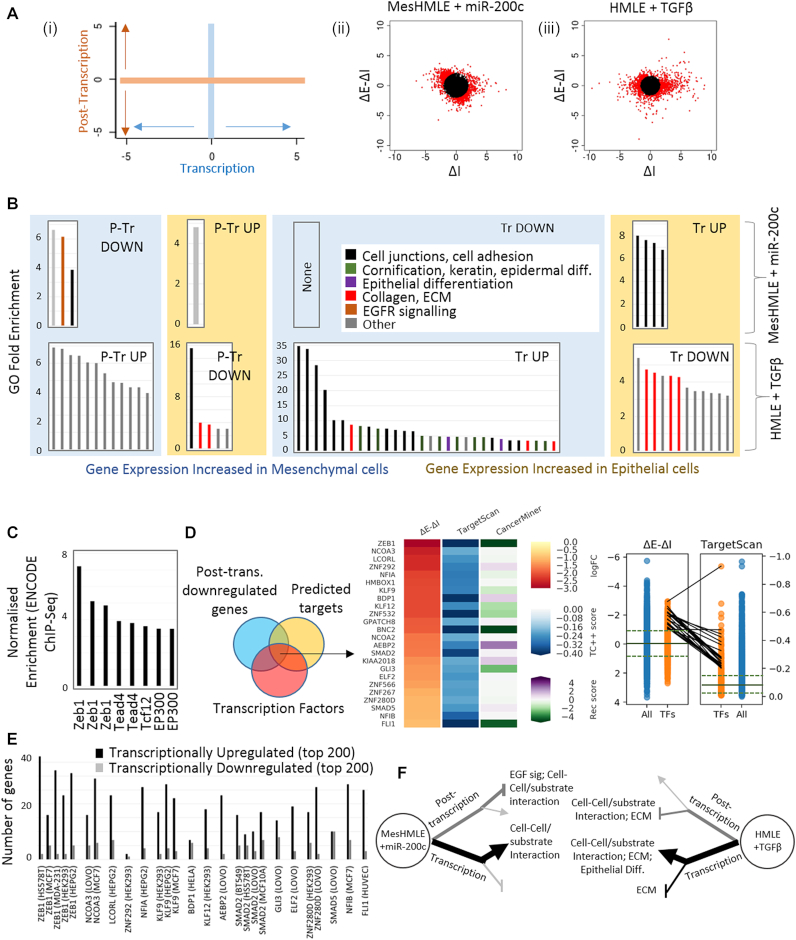
Epithelial-mesenchymal transition (EMT) has been a subject of intense scrutiny as it facilitates metastasis and alters drug sensitivity. Although EMT-regulatory roles for numerous miRNAs and transcription factors are known, their functions can be difficult to disentangle, in part due to the difficulty in identifying direct miRNA targets from complex datasets and in deciding how to incorporate 'indirect' miRNA effects that may, or may not, represent biologically relevant information. To better understand how miRNAs exert effects throughout the transcriptome during EMT, we employed Exon-Intron Split Analysis (EISA), a bioinformatic technique that separates transcriptional and post-transcriptional effects through the separate analysis of RNA-Seq reads mapping to exons and introns. We find that in response to the manipulation of miRNAs, a major effect on gene expression is transcriptional. We also find extensive co-ordination of transcriptional and post-transcriptional regulatory mechanisms during both EMT and mesenchymal to epithelial transition (MET) in response to TGF-β or miR-200c respectively. The prominent transcriptional influence of miRNAs was also observed in other datasets where miRNA levels were perturbed. This work cautions against a narrow approach that is limited to the analysis of direct targets, and demonstrates the utility of EISA to examine complex regulatory networks involving both transcriptional and post-transcriptional mechanisms.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures



References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
