

Assignment of virus and antimicrobial resistance genes to microbial hosts in a complex microbial community by combined long-read assembly and proximity ligation
- PMID: 31375138
- PMCID: PMC6676630
- DOI: 10.1186/s13059-019-1760-x
Assignment of virus and antimicrobial resistance genes to microbial hosts in a complex microbial community by combined long-read assembly and proximity ligation
Abstract
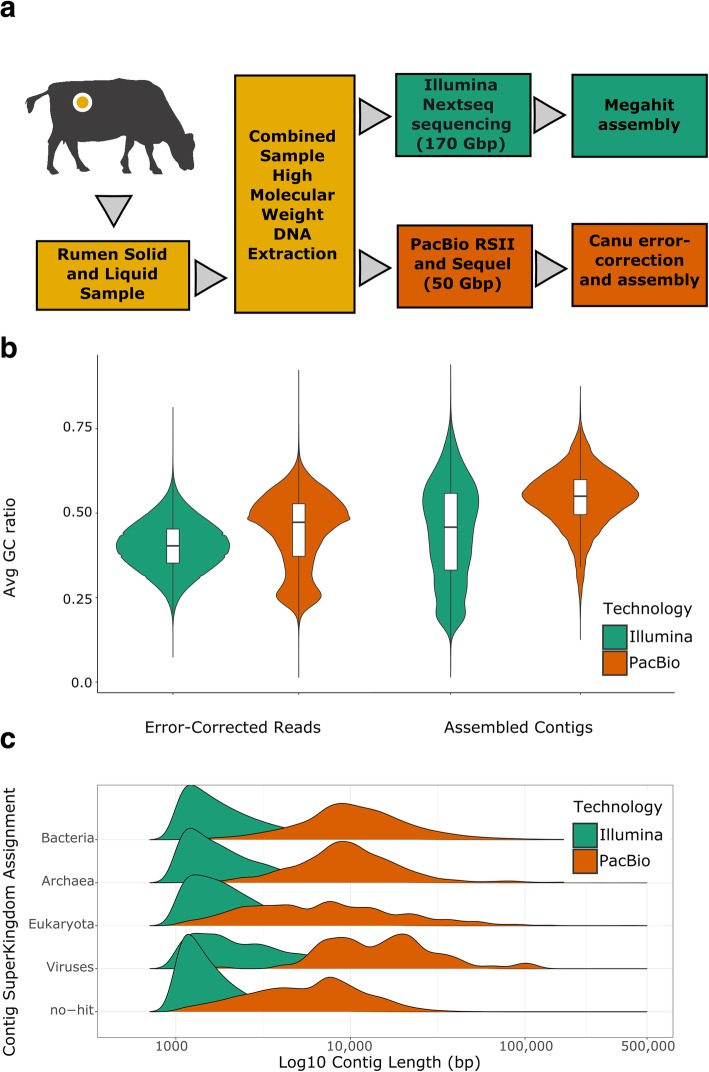
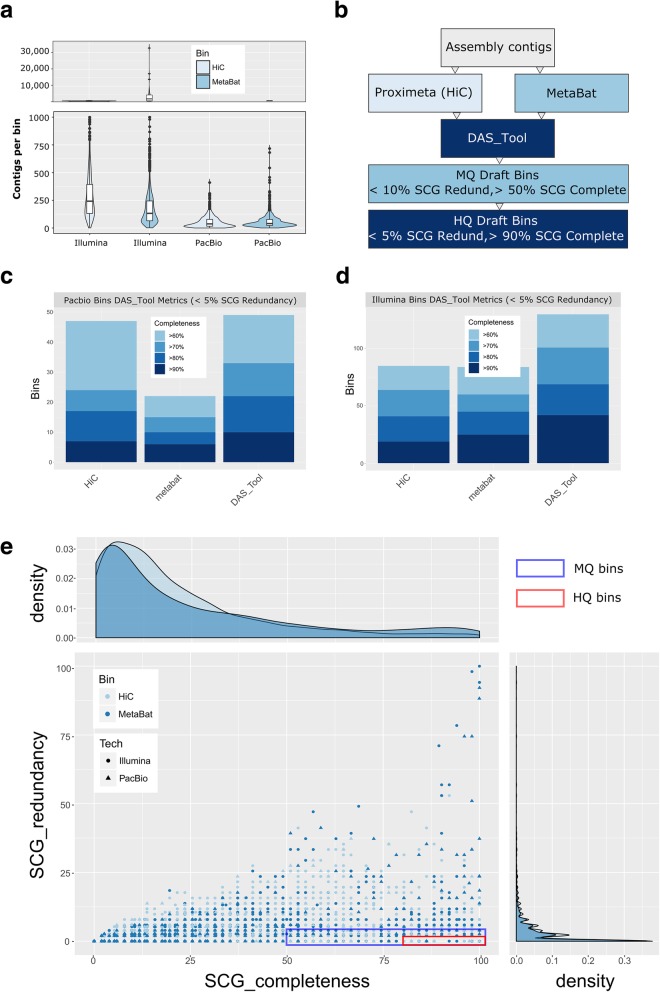
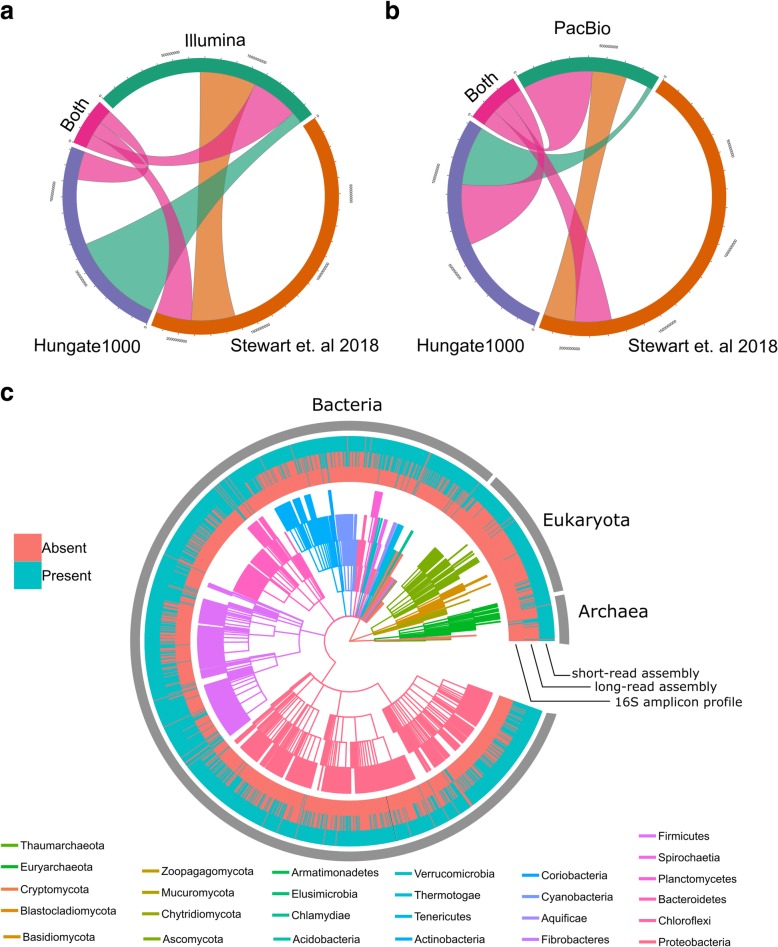
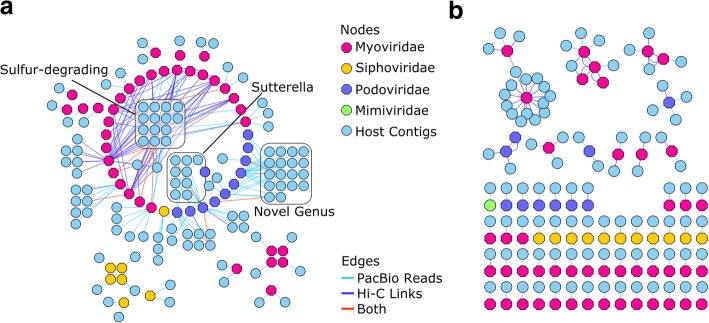
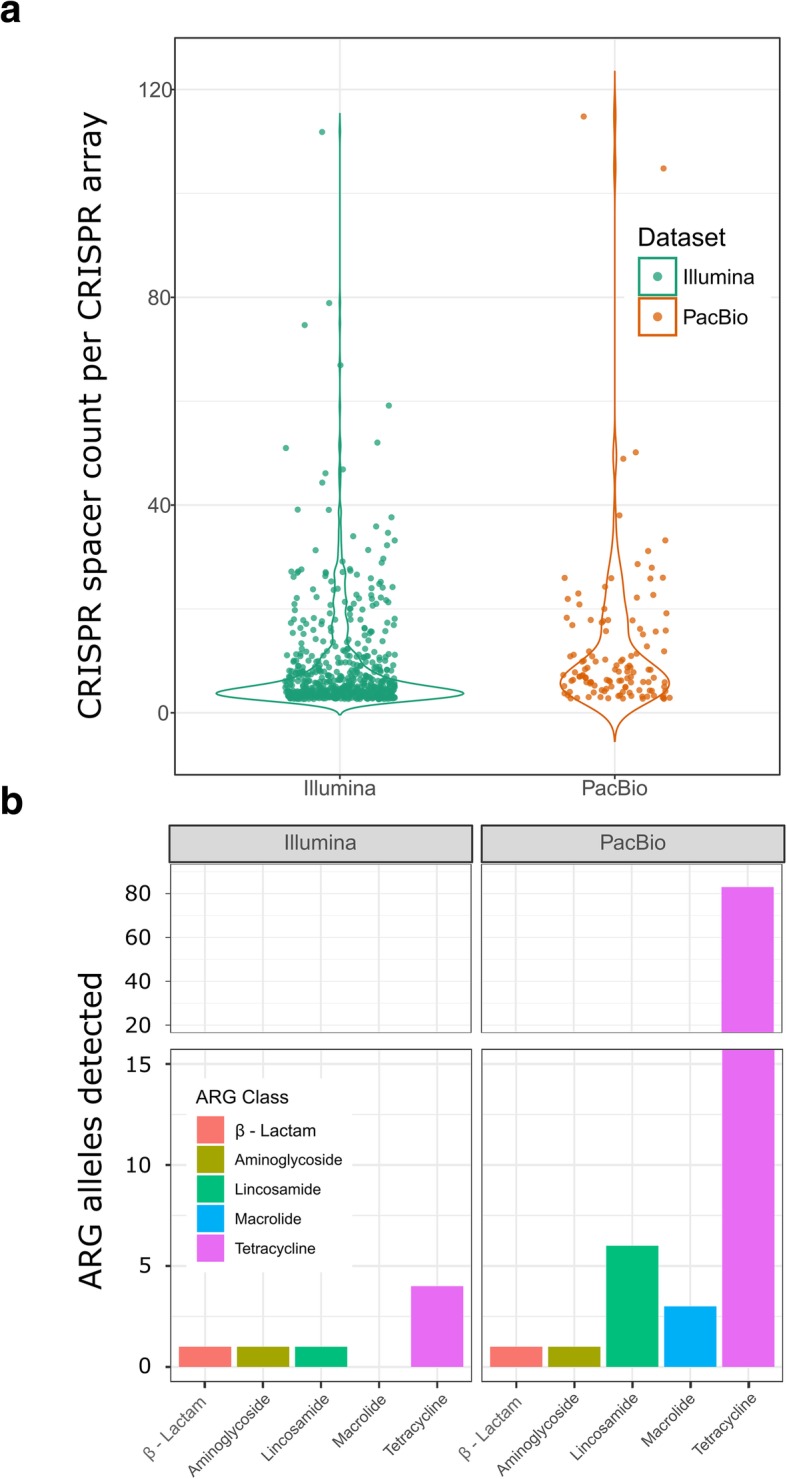
We describe a method that adds long-read sequencing to a mix of technologies used to assemble a highly complex cattle rumen microbial community, and provide a comparison to short read-based methods. Long-read alignments and Hi-C linkage between contigs support the identification of 188 novel virus-host associations and the determination of phage life cycle states in the rumen microbial community. The long-read assembly also identifies 94 antimicrobial resistance genes, compared to only seven alleles in the short-read assembly. We demonstrate novel techniques that work synergistically to improve characterization of biological features in a highly complex rumen microbial community.
Keywords: Hi-C; Metagenome assembly; Metagenomics; PacBio; Virus-host association.
Conflict of interest statement
CH is an employee of Pacific Biosciences. IL, MOP, and STS are employees of Phase Genomics. The other authors declare that they have no competing interests.
Figures
References
-
- Awad S, Irber L, Brown CT. Evaluating metagenome assembly on a simple defined community with many strain variants. bioRxiv. 2017;3:155358.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
