

Functional genetic variants can mediate their regulatory effects through alteration of transcription factor binding
- PMID: 31375681
- PMCID: PMC6677801
- DOI: 10.1038/s41467-019-11412-5
Functional genetic variants can mediate their regulatory effects through alteration of transcription factor binding
Abstract
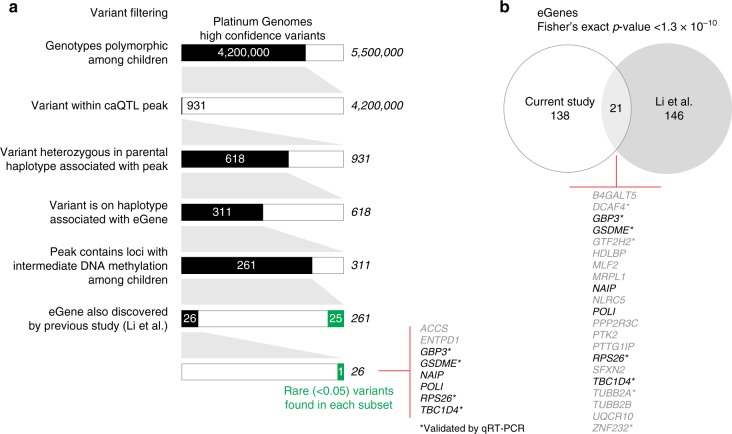
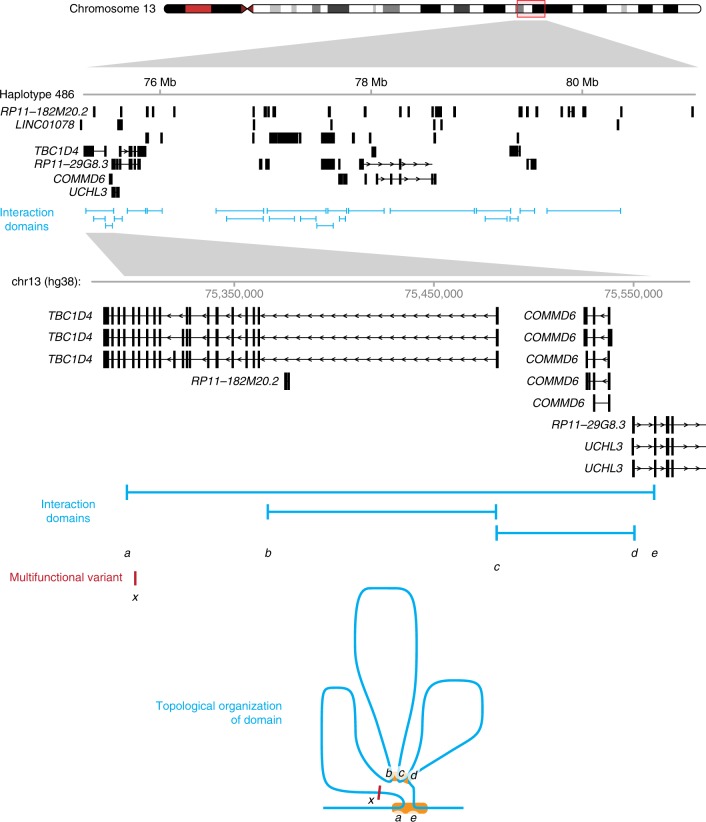
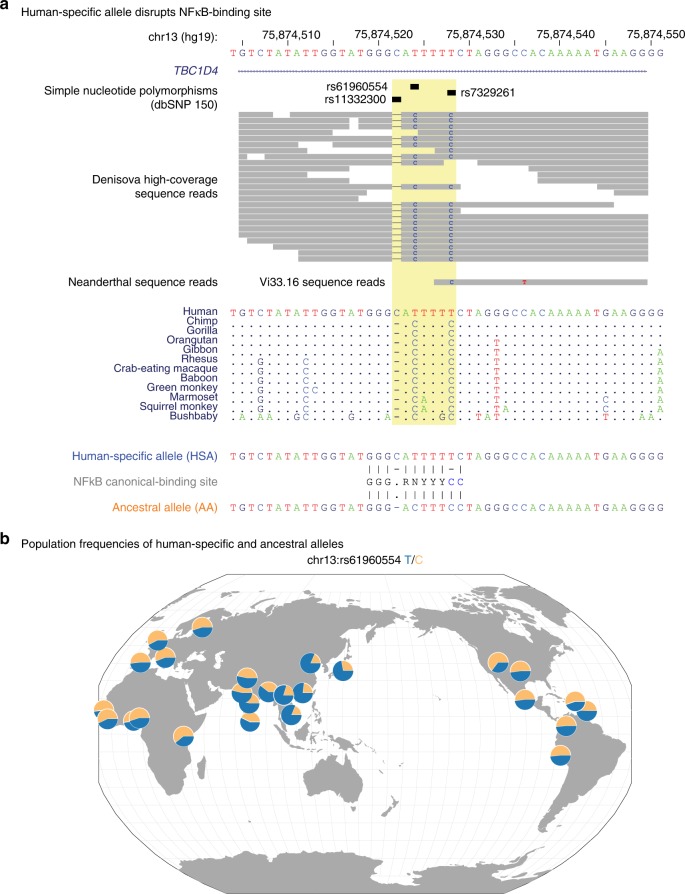
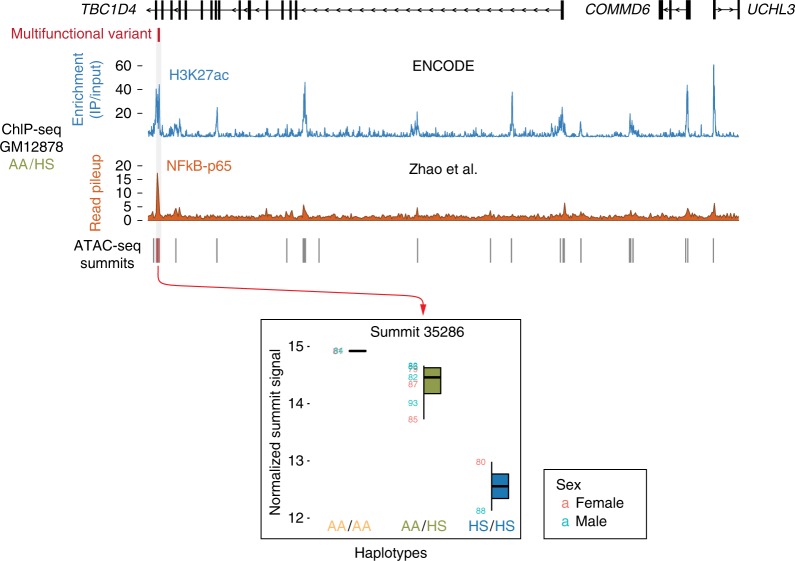
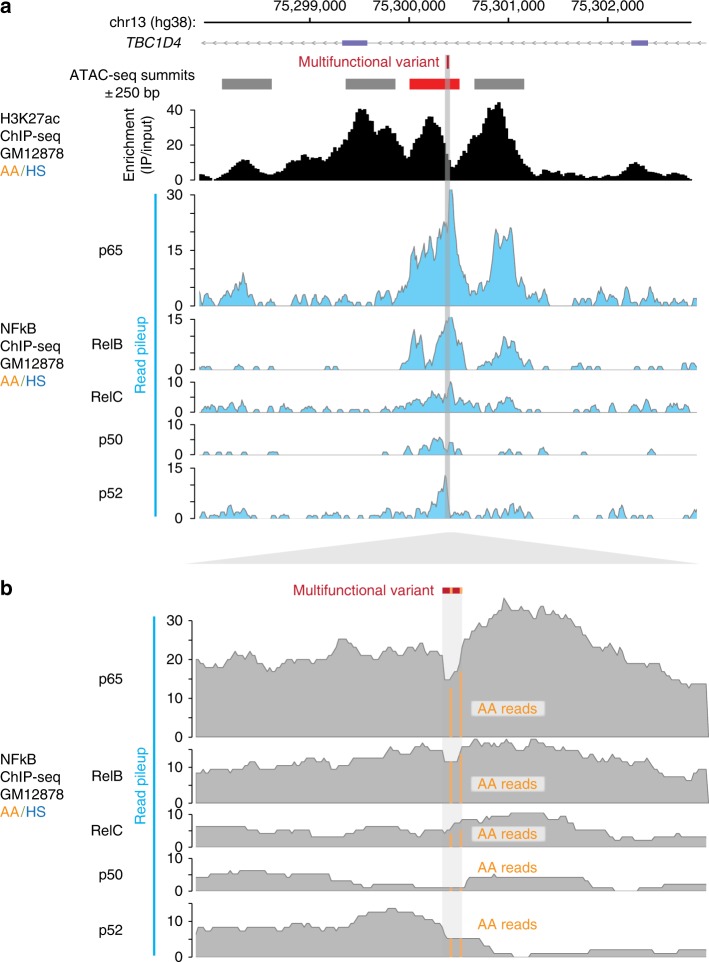
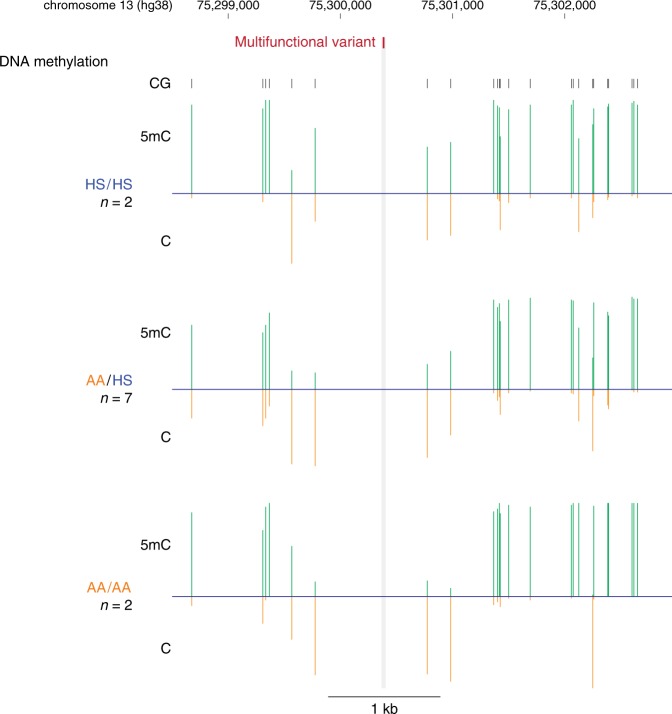
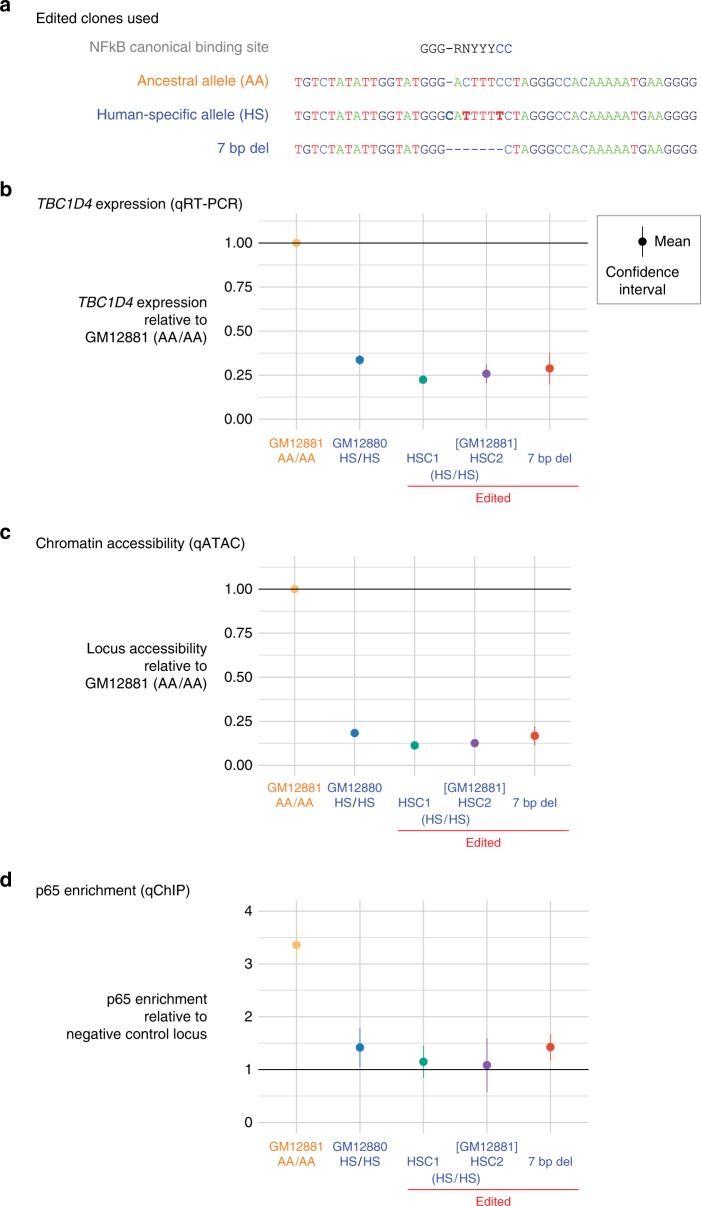
Functional variants in the genome are usually identified by their association with local gene expression, DNA methylation or chromatin states. DNA sequence motif analysis and chromatin immunoprecipitation studies have provided indirect support for the hypothesis that functional variants alter transcription factor binding to exert their effects. In this study, we provide direct evidence that functional variants can alter transcription factor binding. We identify a multifunctional variant within the TBC1D4 gene encoding a canonical NFκB binding site, and edited it using CRISPR-Cas9 to remove this site. We show that this editing reduces TBC1D4 expression, local chromatin accessibility and binding of the p65 component of NFκB. We then used CRISPR without genomic editing to guide p65 back to the edited locus, demonstrating that this re-targeting, occurring ~182 kb from the gene promoter, is enough to restore the function of the locus, supporting the central role of transcription factors mediating the effects of functional variants.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
