

Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype
- PMID: 31375807
- PMCID: PMC7605509
- DOI: 10.1038/s41587-019-0201-4
Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype
Abstract
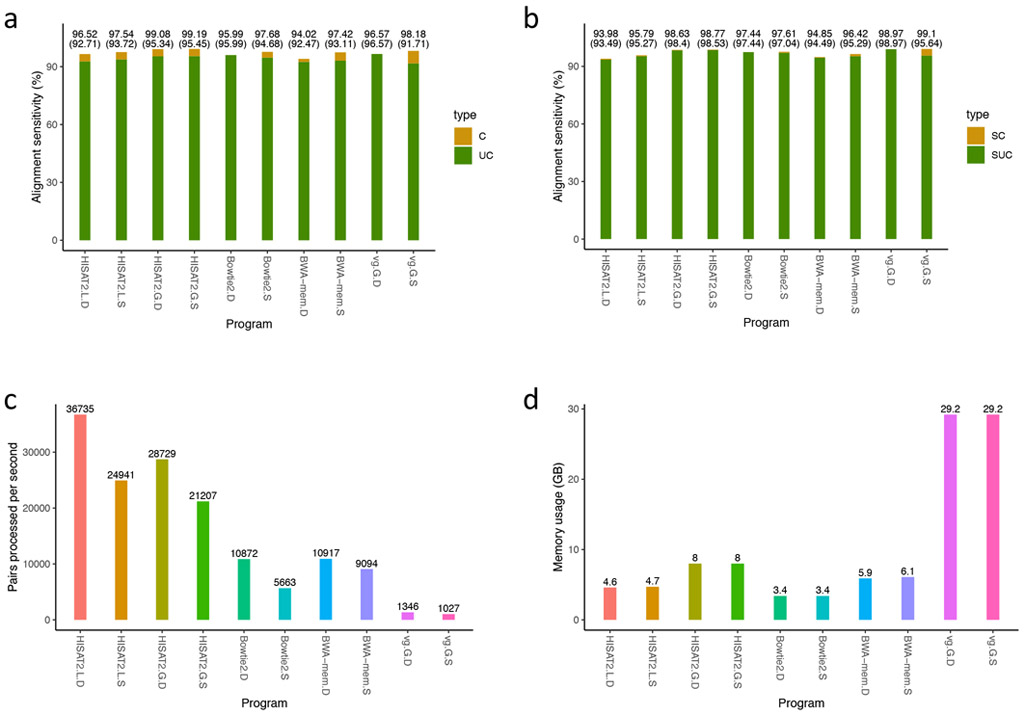
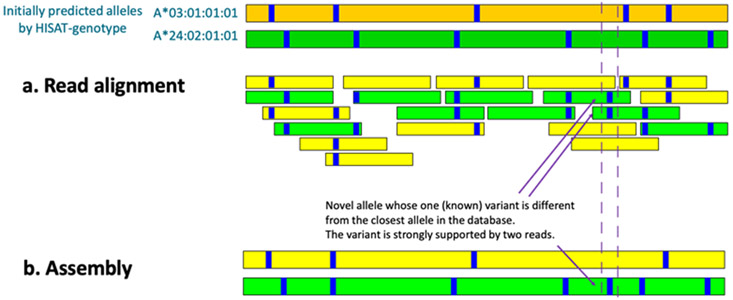
The human reference genome represents only a small number of individuals, which limits its usefulness for genotyping. We present a method named HISAT2 (hierarchical indexing for spliced alignment of transcripts 2) that can align both DNA and RNA sequences using a graph Ferragina Manzini index. We use HISAT2 to represent and search an expanded model of the human reference genome in which over 14.5 million genomic variants in combination with haplotypes are incorporated into the data structure used for searching and alignment. We benchmark HISAT2 using simulated and real datasets to demonstrate that our strategy of representing a population of genomes, together with a fast, memory-efficient search algorithm, provides more detailed and accurate variant analyses than other methods. We apply HISAT2 for HLA typing and DNA fingerprinting; both applications form part of the HISAT-genotype software that enables analysis of haplotype-resolved genes or genomic regions. HISAT-genotype outperforms other computational methods and matches or exceeds the performance of laboratory-based assays.
Figures
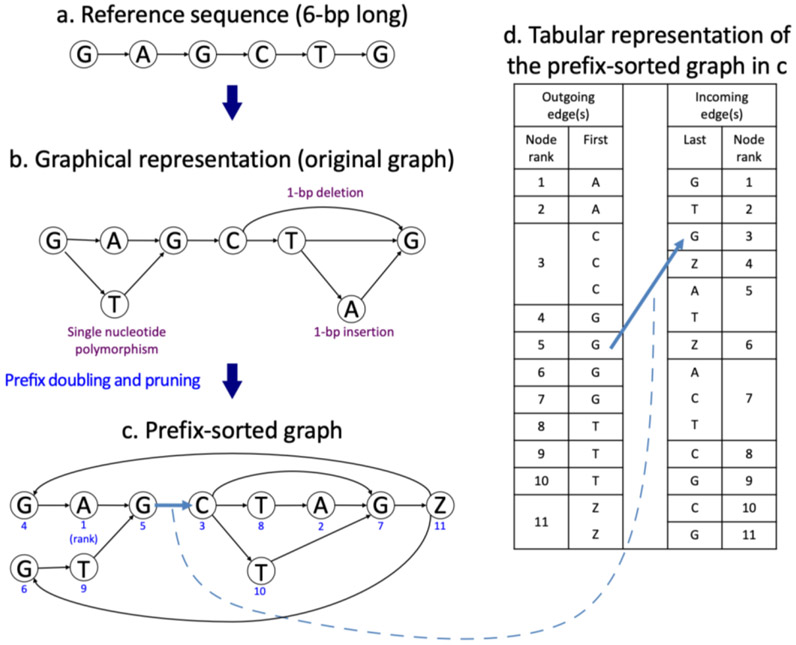
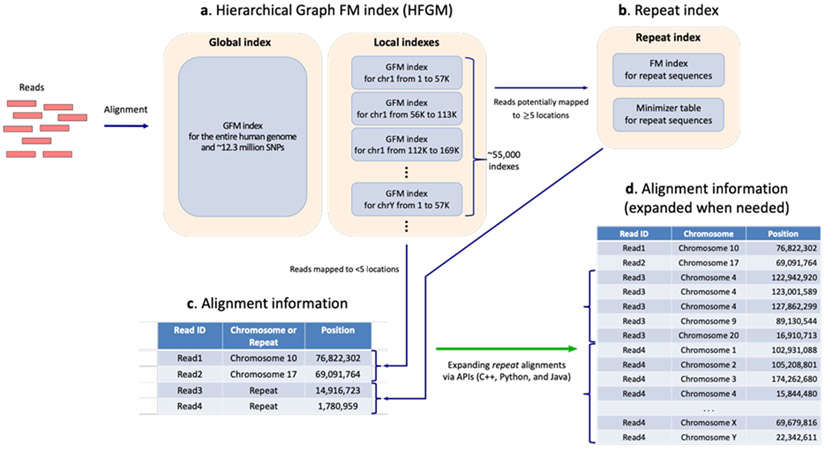
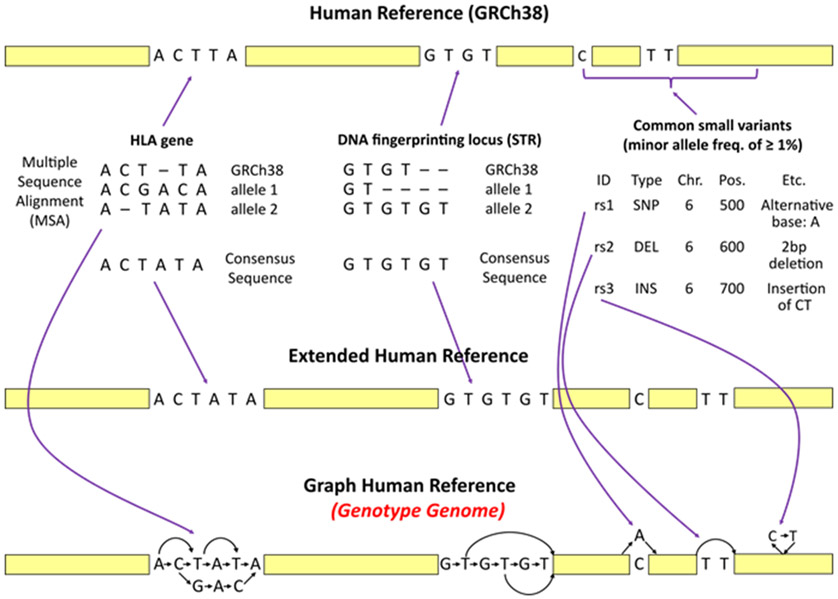
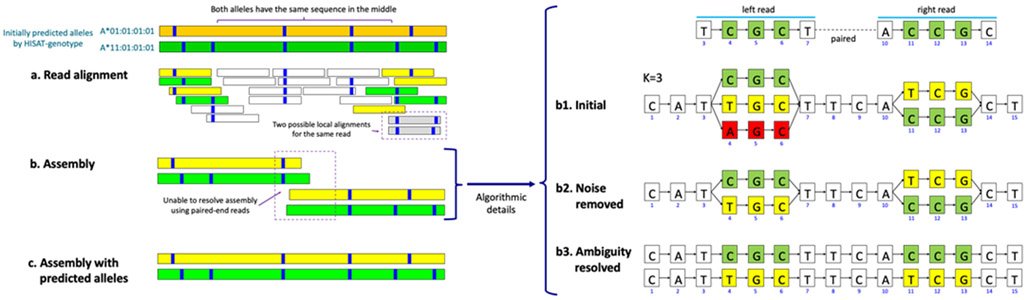
References
-
- t Hoen PA et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nat Biotechnol 31, 1015–1022 (2013). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
