

Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion
- PMID: 31375813
- PMCID: PMC7299161
- DOI: 10.1038/s41587-019-0206-z
Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion
Abstract
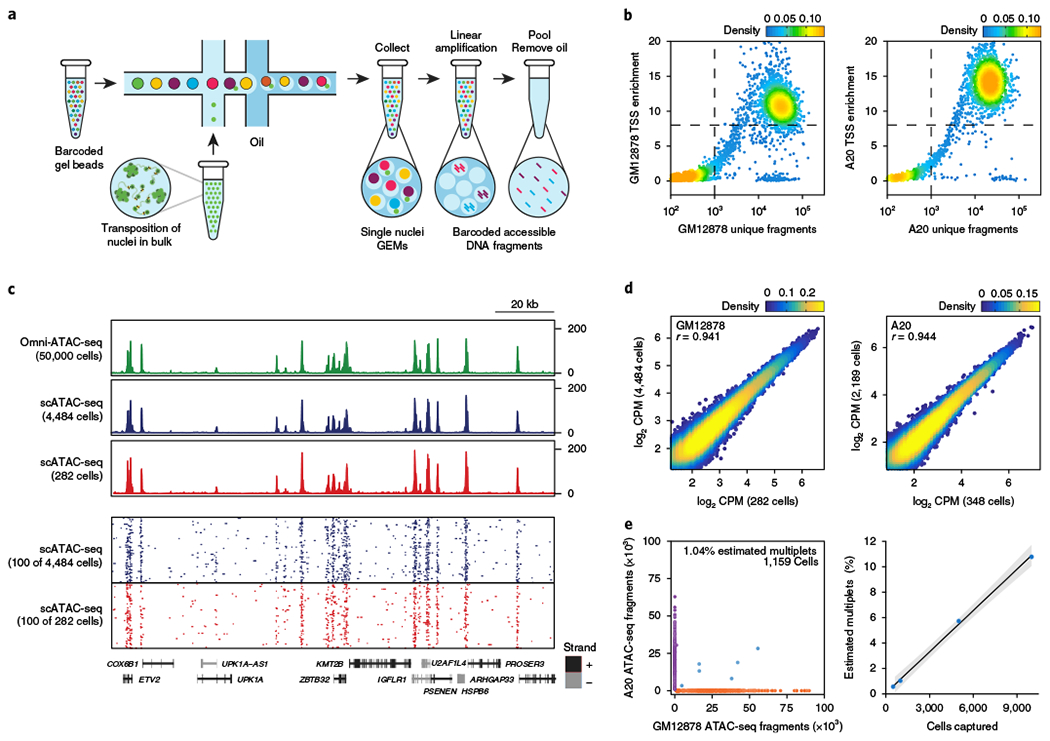
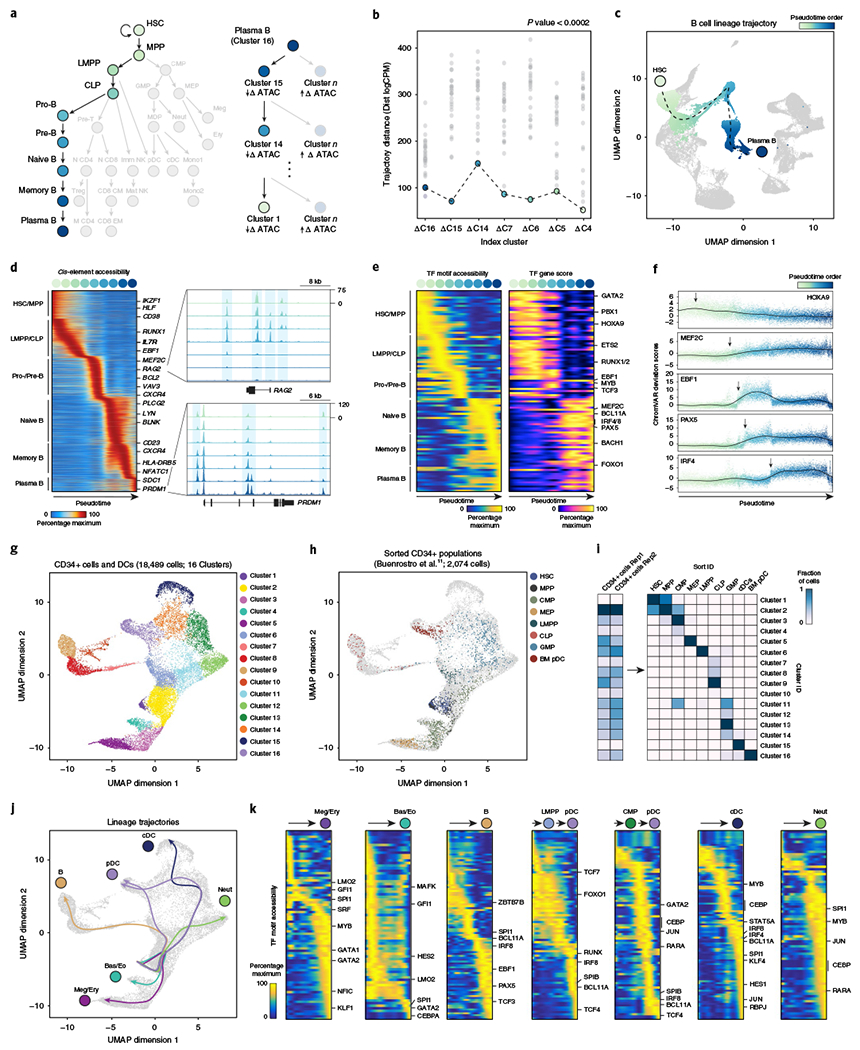
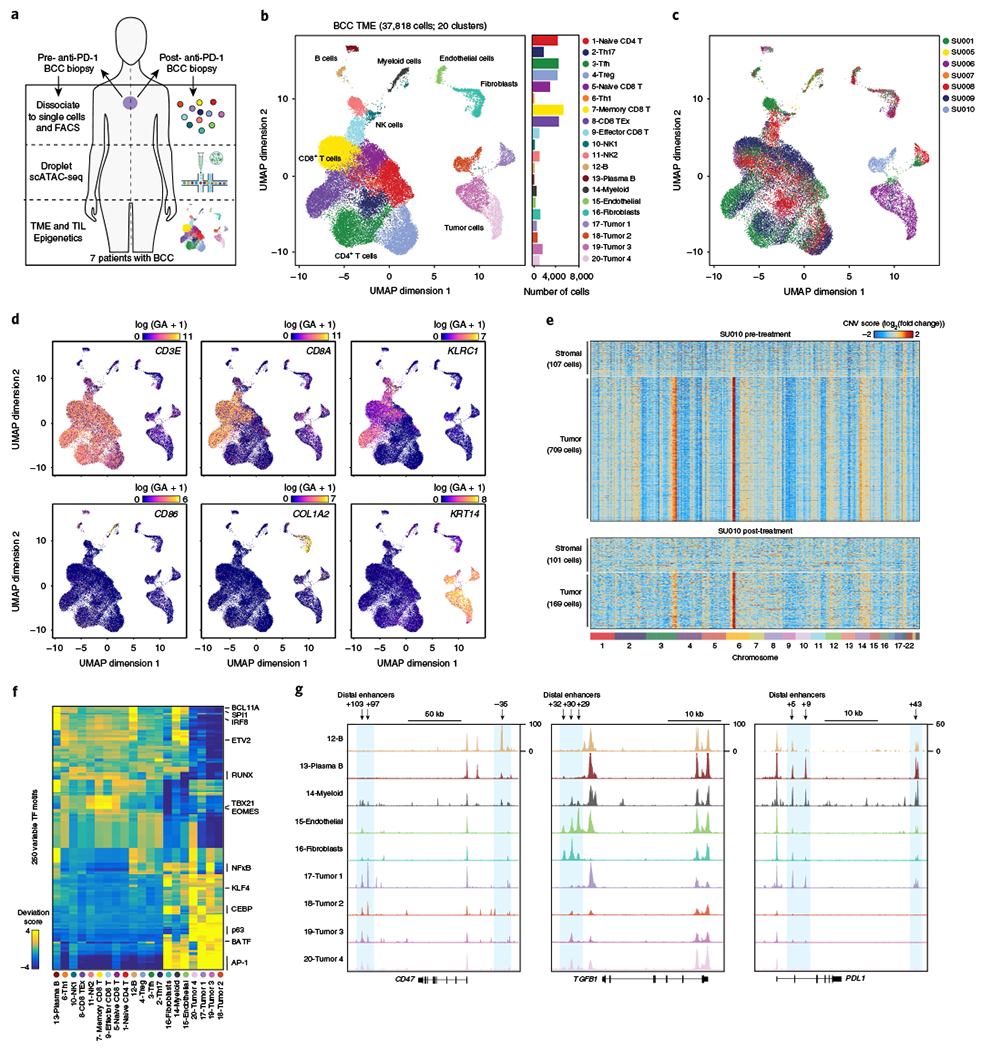
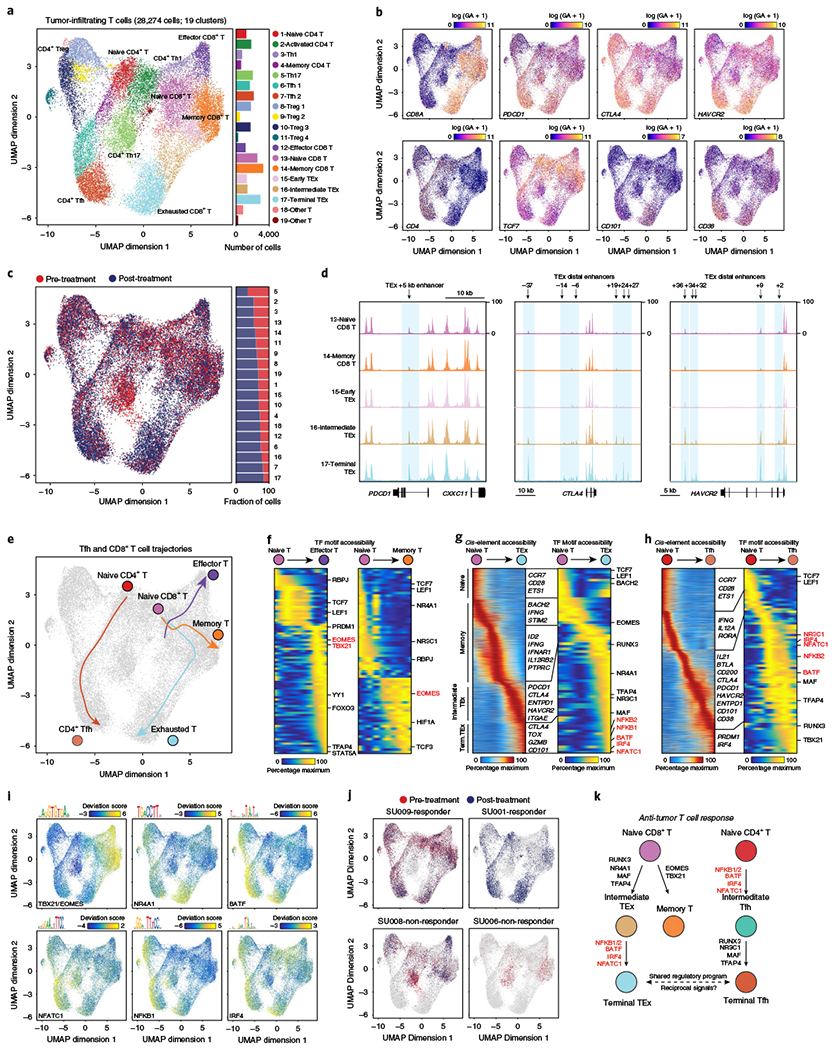
Understanding complex tissues requires single-cell deconstruction of gene regulation with precision and scale. Here, we assess the performance of a massively parallel droplet-based method for mapping transposase-accessible chromatin in single cells using sequencing (scATAC-seq). We apply scATAC-seq to obtain chromatin profiles of more than 200,000 single cells in human blood and basal cell carcinoma. In blood, application of scATAC-seq enables marker-free identification of cell type-specific cis- and trans-regulatory elements, mapping of disease-associated enhancer activity and reconstruction of trajectories of cellular differentiation. In basal cell carcinoma, application of scATAC-seq reveals regulatory networks in malignant, stromal and immune cells in the tumor microenvironment. Analysis of scATAC-seq profiles from serial tumor biopsies before and after programmed cell death protein 1 blockade identifies chromatin regulators of therapy-responsive T cell subsets and reveals a shared regulatory program that governs intratumoral CD8+ T cell exhaustion and CD4+ T follicular helper cell development. We anticipate that scATAC-seq will enable the unbiased discovery of gene regulatory factors across diverse biological systems.
Conflict of interest statement
Competing interests
H.Y.C. is a cofounder of Accent Therapeutics and Epinomics and is an adviser to 10x Genomics and Spring Discovery. W.J.G. is a cofounder of Epinomics and an adviser to 10x Genomics, Guardant Health and Centrillion. A.T.S. is an advisor to Immunai. F.M., G.P.M., B.N.O., P.S., J.C.B., D.J., C.M.N., J.W., L.W., Y.Y., P.G.G. and G.Y.Z. are employees of 10x Genomics. A.L.S.C. was an advisory board member and clinical investigator for studies sponsored by Merck, Regeneron, Novartis, Galderma and Genentech Roche. Stanford University holds patents on ATAC-seq, on which P.G., W.J.G. and H.Y.C. are named as inventors.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
