

1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) Urea, a Selective and Potent Dual Inhibitor of Soluble Epoxide Hydrolase and p38 Kinase Intervenes in Alzheimer's Signaling in Human Nerve Cells
- PMID: 31378059
- PMCID: PMC7028313
- DOI: 10.1021/acschemneuro.9b00271
1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) Urea, a Selective and Potent Dual Inhibitor of Soluble Epoxide Hydrolase and p38 Kinase Intervenes in Alzheimer's Signaling in Human Nerve Cells
Abstract
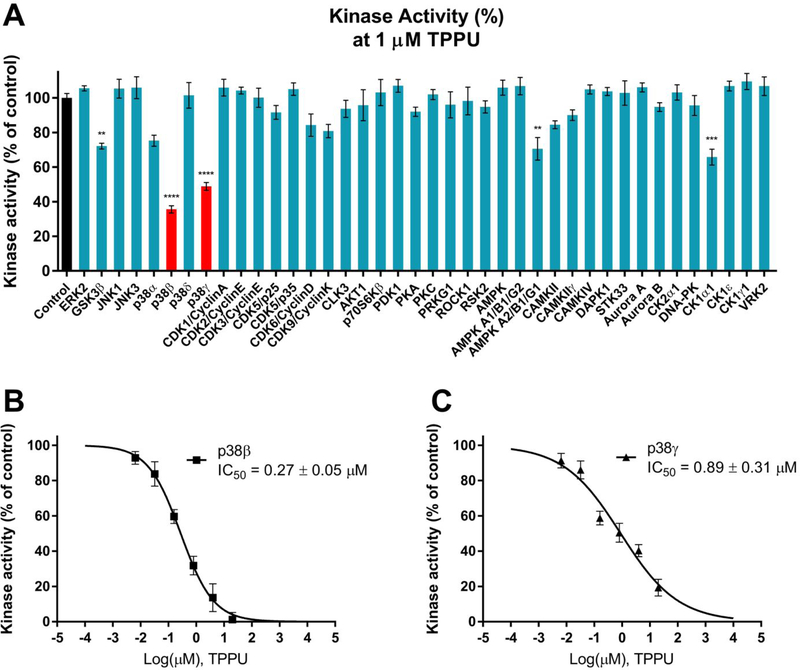
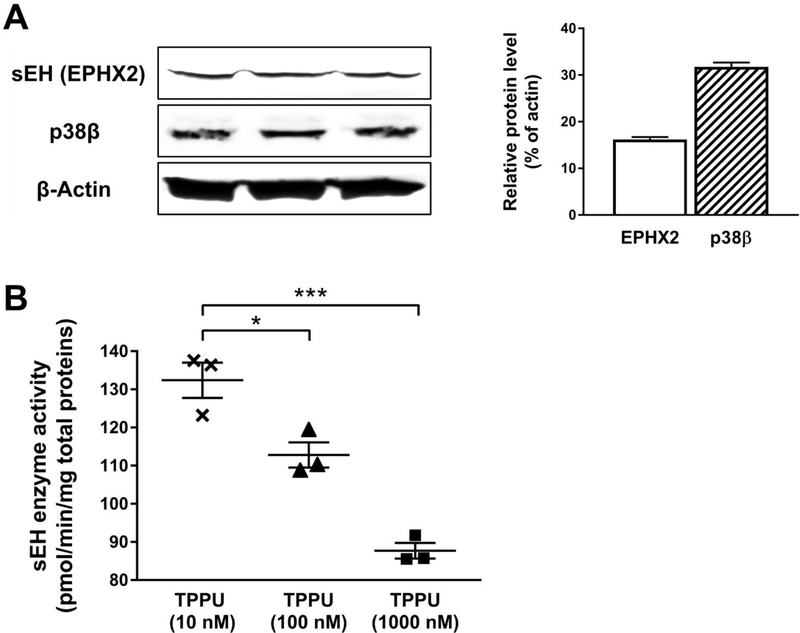
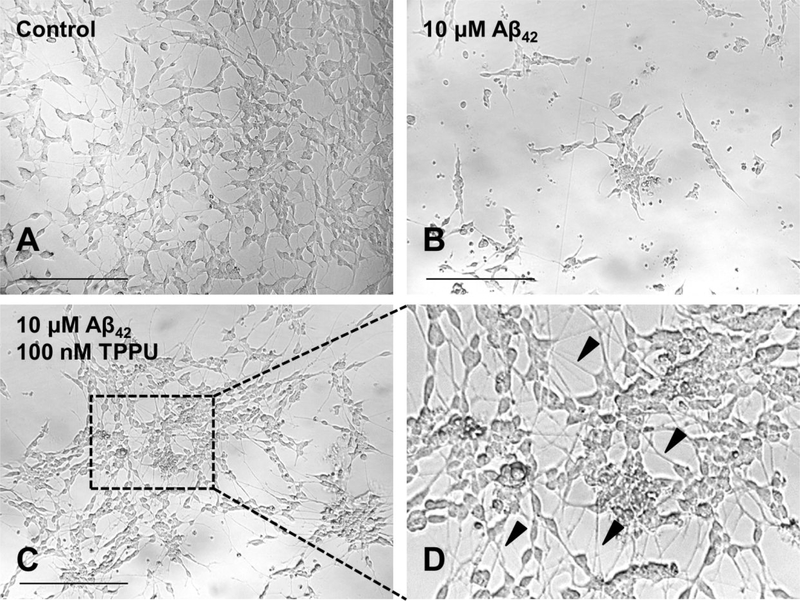
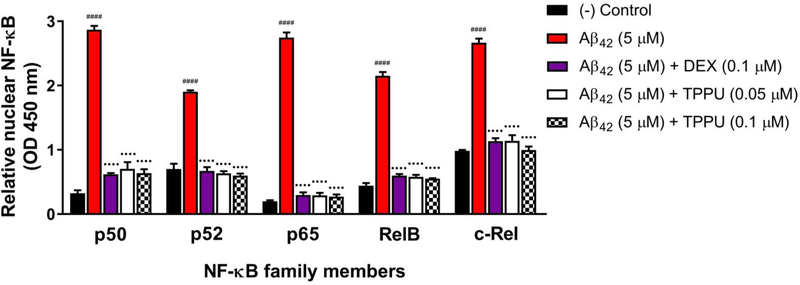
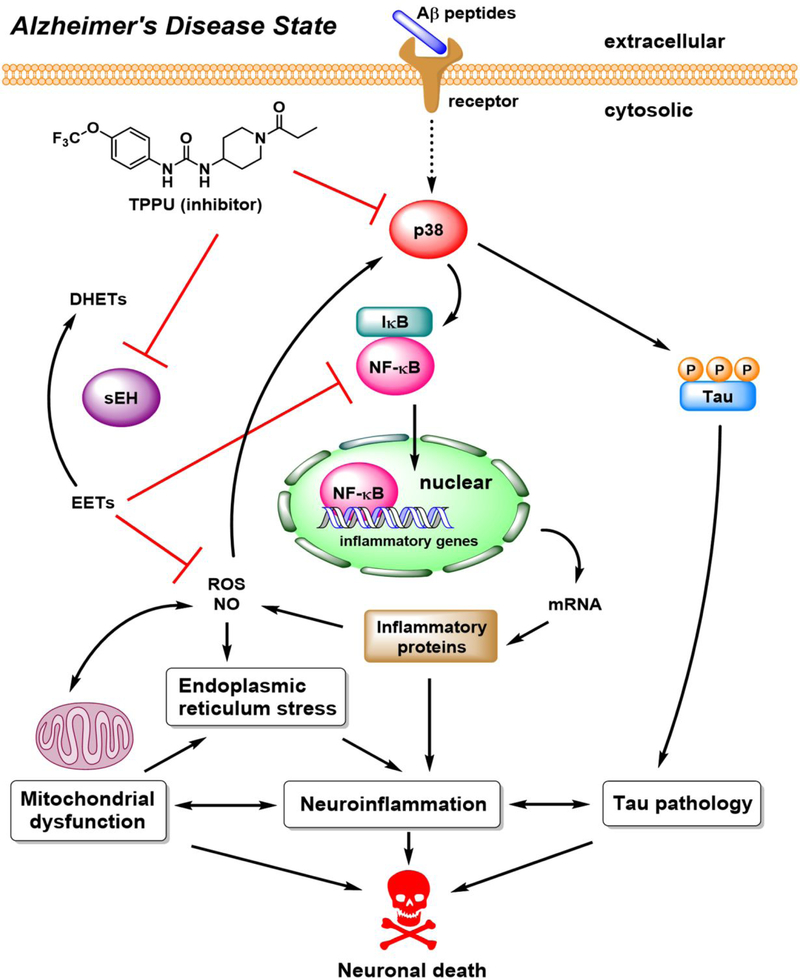
Alzheimer's disease (AD) is the most common neurodegenerative disorder. Neuroinflammation is a prevalent pathogenic stress leading to neuronal death in AD. Targeting neuroinflammation to keep neurons alive is an attractive strategy for AD therapy. 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) is a potent inhibitor of soluble epoxide hydrolase (sEH) and can enter into the brain. It has good efficacy on a wide range of chronic inflammatory diseases in preclinical animal models. However, the anti-neuroinflammatory effects and molecular mechanisms of TPPU for potential AD interventions remain elusive. With an aim to develop multitarget therapeutics for neurodegenerative diseases, we screened TPPU against sEH from different mammalian species and a broad panel of human kinases in vitro for potential new targets relevant to neuroinflammation in AD. TPPU inhibits both human sEH and p38β kinase, two key regulators of inflammation, with nanomolar potencies and distinct selectivity. To further elucidate the molecular mechanisms, differentiated SH-SY5Y human neuroblastoma cells were used as an AD cell model, and we investigated the neuroprotection of TPPU against amyloid oligomers. We found that TPPU effectively prevents neuronal death by mitigating amyloid neurotoxicity, tau hyperphosphorylation, and mitochondrial dysfunction, promoting neurite outgrowth and suppressing activation and nuclear translocation of NF-κB for inflammatory responses in human nerve cells. The results indicate that TPPU is a potent and selective dual inhibitor of sEH and p38β kinase, showing a synergistic action in multiple AD signaling pathways. Our study sheds light upon TPPU and other sEH/p38β dual inhibitors for potential pharmacological interventions in AD.
Keywords: Alzheimer’s disease; dual inhibitor; mitochondrial dysfunction; neuroinflammation; neuroprotection; p38 mitogen-activated protein kinase; soluble epoxide hydrolase.
Conflict of interest statement
Conflict of Interest
B.D.H. is a founder of EicOsis Human Health which is developing sEH inhibitors for treatment of inflammatory and neuropathic pain.
Figures
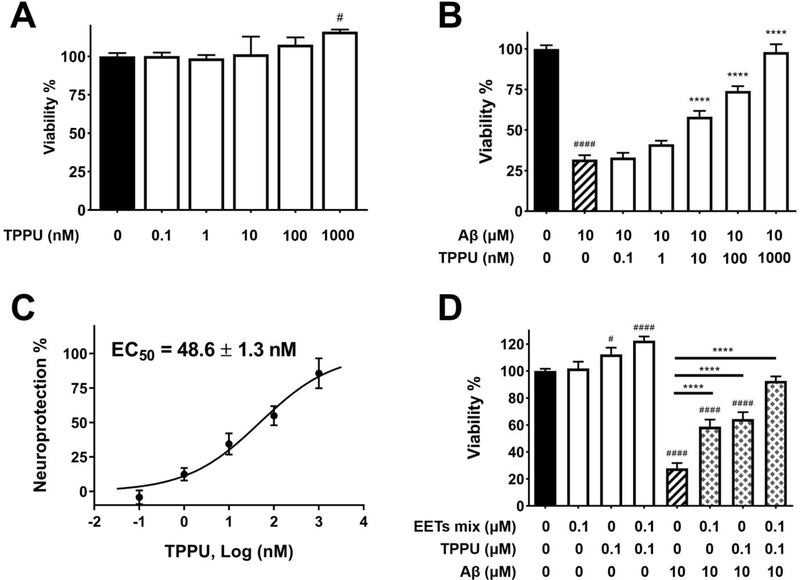
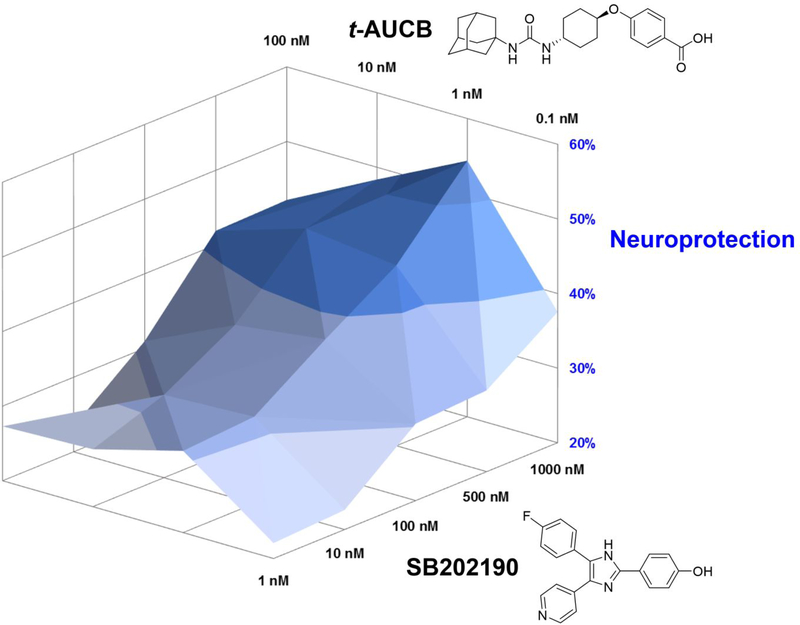
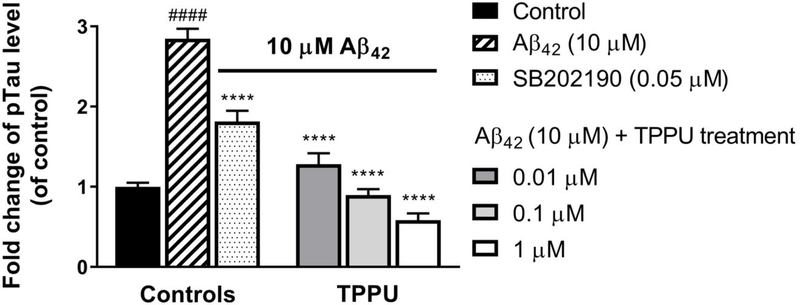
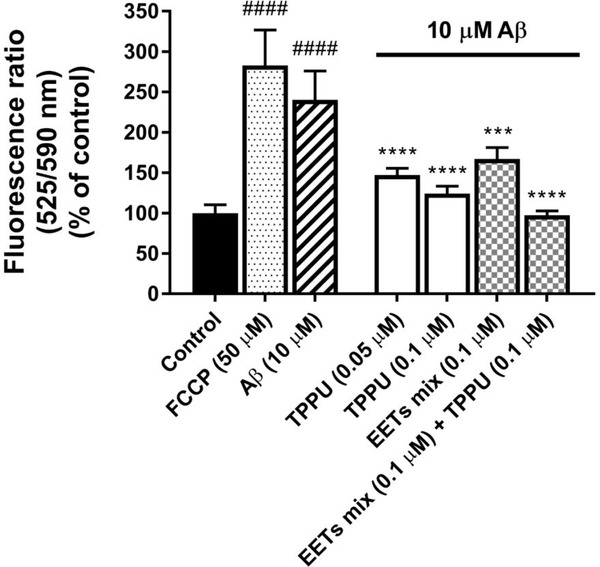
Similar articles
-
A Soluble Epoxide Hydrolase Inhibitor Improves Cerebrovascular Dysfunction, Neuroinflammation, Amyloid Burden, and Cognitive Impairments in the hAPP/PS1 TgF344-AD Rat Model of Alzheimer's Disease.Int J Mol Sci. 2025 Mar 8;26(6):2433. doi: 10.3390/ijms26062433. Int J Mol Sci. 2025. PMID: 40141075 Free PMC article.
-
The soluble epoxide hydrolase inhibitor TPPU alleviates Aβ-mediated neuroinflammatory responses in Drosophila melanogaster and cellular models of alzheimer's disease.J Inflamm (Lond). 2025 Jun 23;22(1):25. doi: 10.1186/s12950-025-00449-7. J Inflamm (Lond). 2025. PMID: 40551105 Free PMC article.
-
A Soluble Epoxide Hydrolase Inhibitor, 1-TrifluoromethoxyPhenyl-3-(1-Propionylpiperidin-4-yl) Urea, Ameliorates Experimental Autoimmune Encephalomyelitis.Int J Mol Sci. 2021 Apr 28;22(9):4650. doi: 10.3390/ijms22094650. Int J Mol Sci. 2021. PMID: 33925035 Free PMC article.
-
Target-Mediated Drug Disposition-A Class Effect of Soluble Epoxide Hydrolase Inhibitors.J Clin Pharmacol. 2021 Apr;61(4):531-537. doi: 10.1002/jcph.1763. Epub 2020 Oct 19. J Clin Pharmacol. 2021. PMID: 33078430 Free PMC article. Review.
-
Soluble epoxide hydrolase as a therapeutic target for pain, inflammatory and neurodegenerative diseases.Pharmacol Ther. 2017 Dec;180:62-76. doi: 10.1016/j.pharmthera.2017.06.006. Epub 2017 Jun 19. Pharmacol Ther. 2017. PMID: 28642117 Free PMC article. Review.
Cited by
-
Genetic deficiency or pharmacological inhibition of soluble epoxide hydrolase ameliorates high fat diet-induced pancreatic β-cell dysfunction and loss.Free Radic Biol Med. 2021 Aug 20;172:48-57. doi: 10.1016/j.freeradbiomed.2021.05.029. Epub 2021 May 24. Free Radic Biol Med. 2021. PMID: 34038767 Free PMC article.
-
Natural Products in the Prevention of Metabolic Diseases: Lessons Learned from the 20th KAST Frontier Scientists Workshop.Nutrients. 2021 May 31;13(6):1881. doi: 10.3390/nu13061881. Nutrients. 2021. PMID: 34072678 Free PMC article. Review.
-
Differential lipid signaling from CD4+ and CD8+ T cells contributes to type 1 diabetes development.Front Immunol. 2024 Sep 18;15:1444639. doi: 10.3389/fimmu.2024.1444639. eCollection 2024. Front Immunol. 2024. PMID: 39359722 Free PMC article.
-
Small Molecule Soluble Epoxide Hydrolase Inhibitors in Multitarget and Combination Therapies for Inflammation and Cancer.Molecules. 2020 Nov 24;25(23):5488. doi: 10.3390/molecules25235488. Molecules. 2020. PMID: 33255197 Free PMC article. Review.
-
1,3-Dichloroadamantyl-Containing Ureas as Potential Triple Inhibitors of Soluble Epoxide Hydrolase, p38 MAPK and c-Raf.Int J Mol Sci. 2023 Dec 26;25(1):338. doi: 10.3390/ijms25010338. Int J Mol Sci. 2023. PMID: 38203510 Free PMC article.
References
-
- Lin MT, and Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases, Nature 443, 787–795. - PubMed
-
- Griffin WST (2013) Neuroinflammatory cytokine signaling and Alzheimer’s disease, N. Engl. J. Med 368, 770–771. - PubMed
-
- Chu D, and Liu F. (2019) Pathological changes of tau related to Alzheimer’s disease, ACS Chem. Neurosci 10, 931–944. - PubMed
-
- Oset-Gasque MJ, and Marco-Contelles J. (2018) Alzheimer’s disease, the “one-molecule, one-target” paradigm, and the multitarget directed ligand approach, ACS Chem. Neurosci 9, 401–403. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
