

Proband Whole-Exome Sequencing Identified Genes Responsible for Autosomal Recessive Non-Syndromic Hearing Loss in 33 Chinese Nuclear Families
- PMID: 31379920
- PMCID: PMC6650584
- DOI: 10.3389/fgene.2019.00639
Proband Whole-Exome Sequencing Identified Genes Responsible for Autosomal Recessive Non-Syndromic Hearing Loss in 33 Chinese Nuclear Families
Abstract
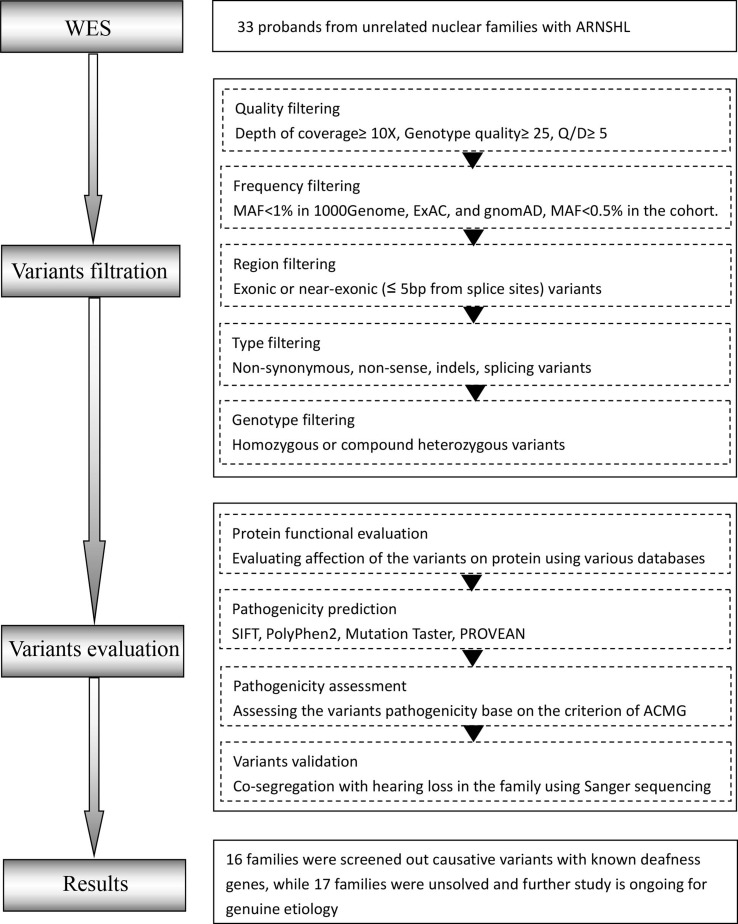
Autosomal recessive non-syndromic hearing loss (ARNSHL) is a highly heterogeneous disease involving more than 70 pathogenic genes. However, most ARNSHL families have small-sized pedigrees with limited genetic information, rendering challenges for the molecular diagnosis of these patients. Therefore, we attempted to establish a strategy for identifying deleterious variants associated with ARNSHL by applying proband whole-exome sequencing (proband-WES). Aside from desiring to improve molecular diagnostic rates, we also aimed to search for novel deafness genes shared by patients with similar phenotype, making up for the deficiency of small ARNSHL families. In this study, 48.5% (16/33) families were detected the pathogenic variants in eight known deafness genes, including 10 novel variants identified in TMPRSS3 (MIM 605551), MYO15A (MIM 602666), TMC1 (MIM 606706), ADGRV1 (MIM 602851), and PTPRQ (MIM 603317). Apart from six novel variants with a truncating effect (nonsense, deletion, insertion, and splice-site), four novel missense variants were not found in 200 unrelated control population by using Sanger sequencing. It is important to note that none of novel genes were shared across different pedigrees, indicating that a larger sample size might be needed. Proband-WES is a cost-effective and precise way of identifying causative variants in nuclear families with ARNSHL. This economical strategy may be appropriated as a clinical application to provide molecular diagnostics, genetic counseling, and individualized health maintenance measures for patients with ARNSHL at hearing clinics.
Keywords: ARNSHL; molecular diagnosis; nuclear families; proband-WES; similar phenotype.
Figures


References
LinkOut - more resources
Full Text Sources