

What lipodystrophies teach us about the metabolic syndrome
- PMID: 31380809
- PMCID: PMC6763226
- DOI: 10.1172/JCI129190
What lipodystrophies teach us about the metabolic syndrome
Abstract
Lipodystrophies are the result of a range of inherited and acquired causes, but all are characterized by perturbations in white adipose tissue function and, in many instances, its mass or distribution. Though patients are often nonobese, they typically manifest a severe form of the metabolic syndrome, highlighting the importance of white fat in the "safe" storage of surplus energy. Understanding the molecular pathophysiology of congenital lipodystrophies has yielded useful insights into the biology of adipocytes and informed therapeutic strategies. More recently, genome-wide association studies focused on insulin resistance have linked common variants to genes implicated in adipose biology and suggested that subtle forms of lipodystrophy contribute to cardiometabolic disease risk at a population level. These observations underpin the use of aligned treatment strategies in insulin-resistant obese and lipodystrophic patients, the major goal being to alleviate the energetic burden on adipose tissue.
Conflict of interest statement
Figures
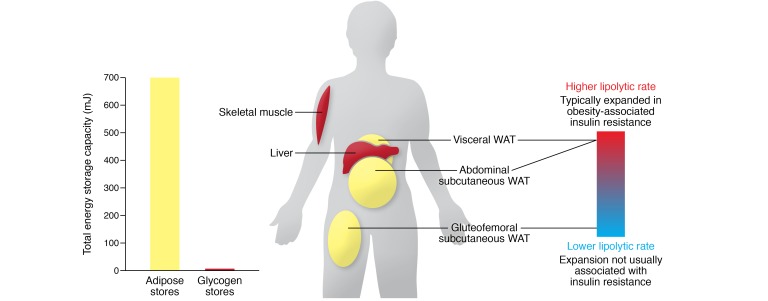
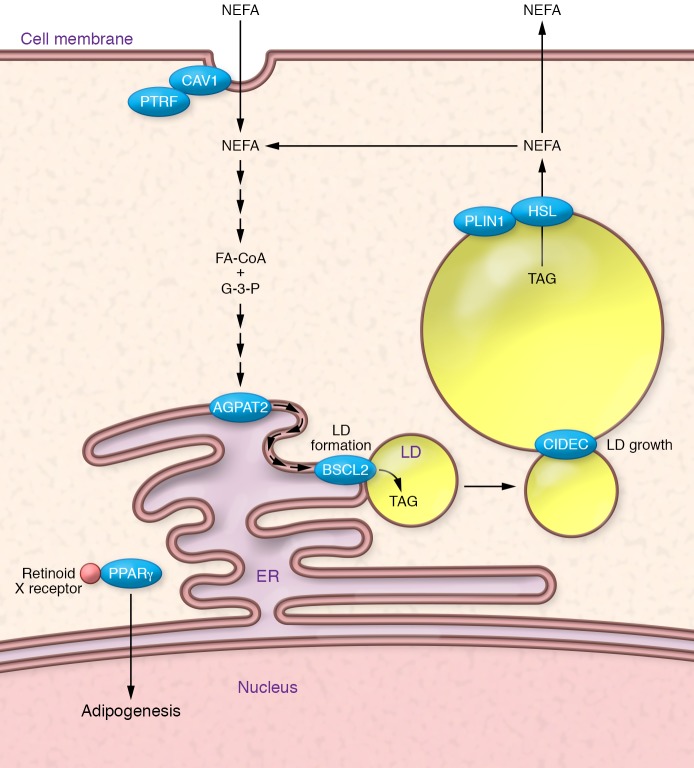
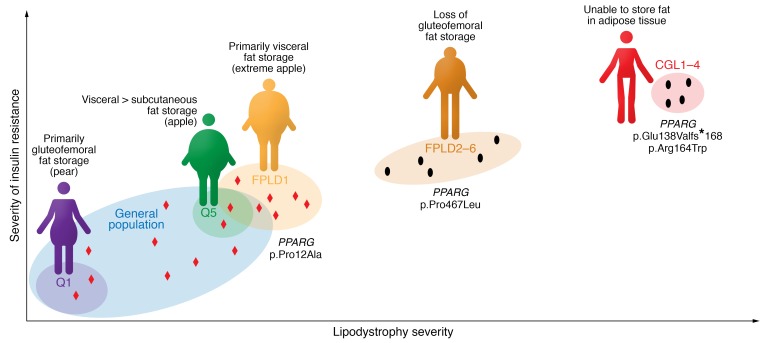
References
-
- Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome — an allostatic perspective. Biochim Biophys Acta. 2010;1801(3):338–349. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
