

Characterization of Neurodevelopmental Abnormalities in iPSC-Derived Striatal Cultures from Patients with Huntington's Disease
- PMID: 31381521
- PMCID: PMC6839479
- DOI: 10.3233/JHD-180333
Characterization of Neurodevelopmental Abnormalities in iPSC-Derived Striatal Cultures from Patients with Huntington's Disease
Abstract
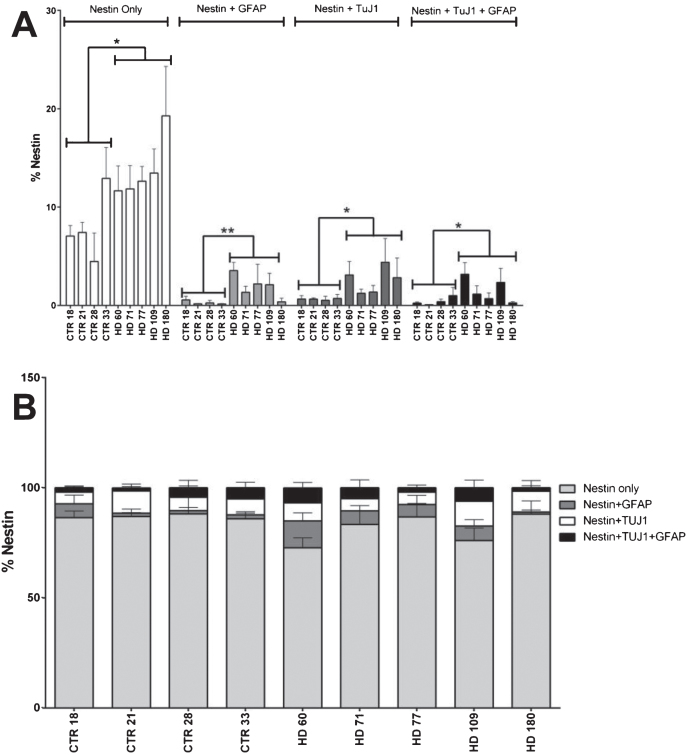
Background: Huntington's disease (HD) is an inherited neurodegenerative disease and is characterized by atrophy of certain regions of the brain in a progressive manner. HD patients experience behavioral changes and uncontrolled movements which can be primarily attributed to the atrophy of striatal neurons. Previous publications describe the models of the HD striatum using induced pluripotent stem cells (iPSCs) derived from HD patients with a juvenile onset (JHD). In this model, the JHD iPSC-derived striatal cultures had altered neurodevelopment and contained a high number of nestin expressing progenitor cells at 42 days of differentiation.
Objective: To further characterize the altered neurodevelopmental phenotype and evaluate potential phenotypic reversal.
Methods: Differentiation of human iPSCs towards striatal fate and characterization by means of immunocytochemistry and stereological quantification.
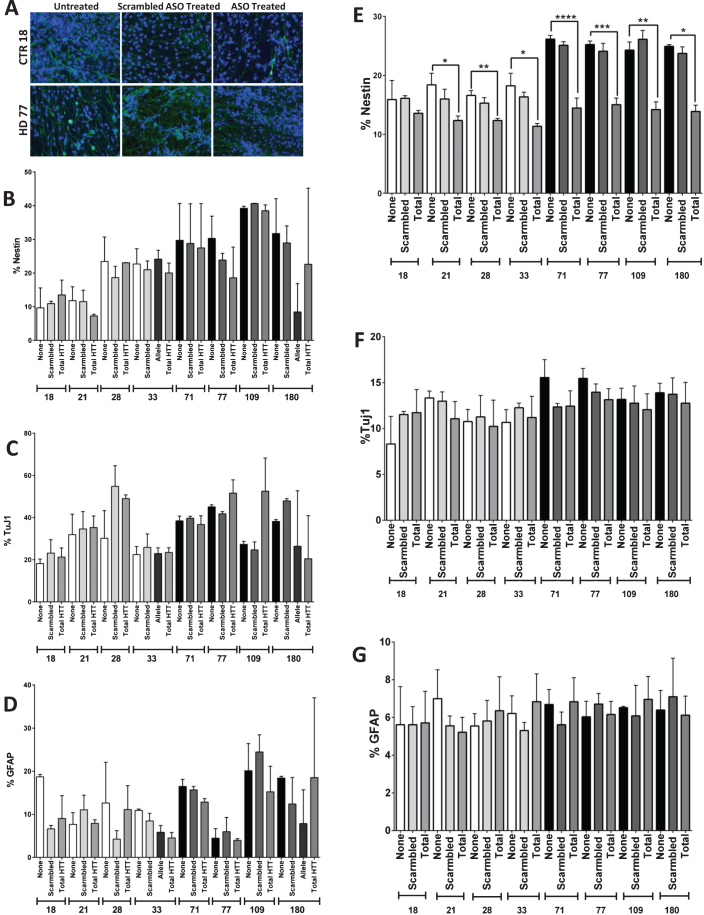
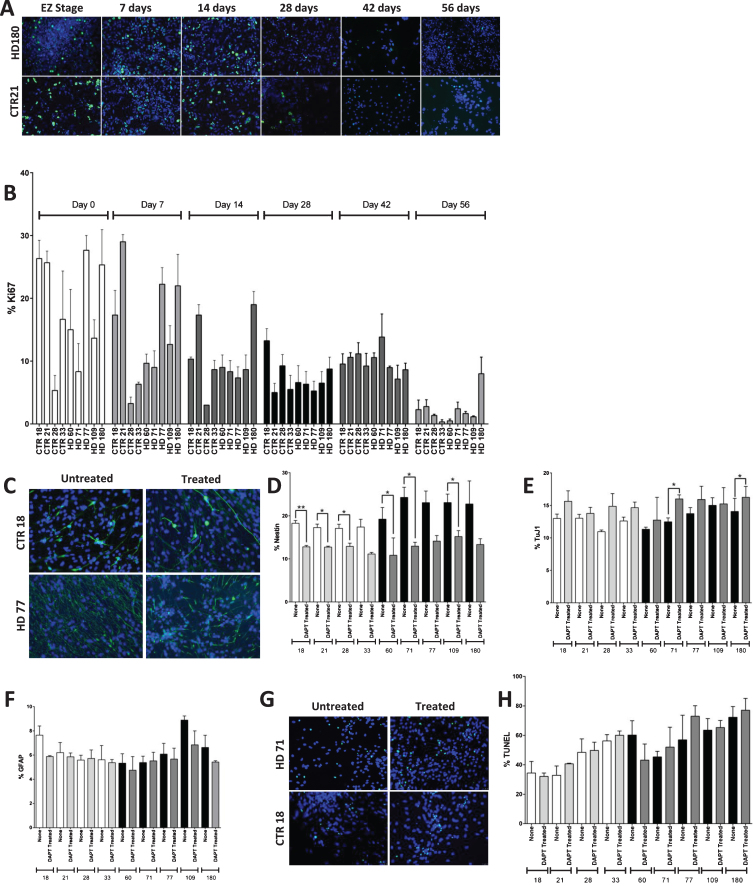
Results: Here this study demonstrates a distinct delay in the differentiation of the JHD neural progenitor population. However, reduction of the JHD aberrant progenitor populations can be accomplished either by targeting the canonical Notch signaling pathway or by treatment with HTT antisense oligonucleotides (ASOs).
Conclusions: In summary, this data is postulated to reflect a potential overall developmental delay in JHD.
Keywords: Huntington’s disease; huntingtin; iPSC; nestin; neurodegenerative disease.
Conflict of interest statement
The authors have no conflict of interest to report.
Figures

Similar articles
-
HD iPSC-derived neural progenitors accumulate in culture and are susceptible to BDNF withdrawal due to glutamate toxicity.Hum Mol Genet. 2015 Jun 1;24(11):3257-71. doi: 10.1093/hmg/ddv080. Epub 2015 Mar 3. Hum Mol Genet. 2015. PMID: 25740845 Free PMC article.
-
Amelioration of Huntington's disease phenotype in astrocytes derived from iPSC-derived neural progenitor cells of Huntington's disease monkeys.PLoS One. 2019 Mar 21;14(3):e0214156. doi: 10.1371/journal.pone.0214156. eCollection 2019. PLoS One. 2019. PMID: 30897183 Free PMC article.
-
Human Huntington's disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species.J Neuroinflammation. 2021 Apr 19;18(1):94. doi: 10.1186/s12974-021-02147-6. J Neuroinflammation. 2021. PMID: 33874957 Free PMC article.
-
Cellular Models: HD Patient-Derived Pluripotent Stem Cells.Methods Mol Biol. 2018;1780:41-73. doi: 10.1007/978-1-4939-7825-0_4. Methods Mol Biol. 2018. PMID: 29856014 Review.
-
Disrupted striatal neuron inputs and outputs in Huntington's disease.CNS Neurosci Ther. 2018 Apr;24(4):250-280. doi: 10.1111/cns.12844. CNS Neurosci Ther. 2018. PMID: 29582587 Free PMC article. Review.
Cited by
-
Molecular Components of Store-Operated Calcium Channels in the Regulation of Neural Stem Cell Physiology, Neurogenesis, and the Pathology of Huntington's Disease.Front Cell Dev Biol. 2021 Apr 1;9:657337. doi: 10.3389/fcell.2021.657337. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33869222 Free PMC article. Review.
-
Huntington's disease iPSC models-using human patient cells to understand the pathology caused by expanded CAG repeats.Fac Rev. 2022 Jun 28;11:16. doi: 10.12703/r/11-16. eCollection 2022. Fac Rev. 2022. PMID: 35865413 Free PMC article. Review.
-
Multi-lineage Human iPSC-Derived Platforms for Disease Modeling and Drug Discovery.Cell Stem Cell. 2020 Mar 5;26(3):309-329. doi: 10.1016/j.stem.2020.02.011. Cell Stem Cell. 2020. PMID: 32142662 Free PMC article. Review.
-
Patient-Specific iPSCs-Based Models of Neurodegenerative Diseases: Focus on Aberrant Calcium Signaling.Int J Mol Sci. 2022 Jan 6;23(2):624. doi: 10.3390/ijms23020624. Int J Mol Sci. 2022. PMID: 35054808 Free PMC article. Review.
-
Transcriptional vulnerabilities of striatal neurons in human and rodent models of Huntington's disease.Nat Commun. 2023 Jan 17;14(1):282. doi: 10.1038/s41467-022-35752-x. Nat Commun. 2023. PMID: 36650127 Free PMC article.
References
-
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–16. - PubMed
-
- Walker FO. Huntington’s disease. Lancet. 2007;369(9557):218–28. - PubMed
-
- van Duijn E, Kingma EM, van der Mast RC. Psychopathology in verified Huntington’s disease gene carriers. J Neuropsychiatry Clin Neurosci. 2007;19(4):441–8. - PubMed
-
- Wild EJ, Tabrizi SJ. Huntington’s disease phenocopy syndromes. Curr Opin Neurol. 2007;20(6):681–7. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
