

PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation
- PMID: 31383640
- PMCID: PMC6776795
- DOI: 10.1182/blood.2019000578
PHF19 promotes multiple myeloma tumorigenicity through PRC2 activation and broad H3K27me3 domain formation
Abstract
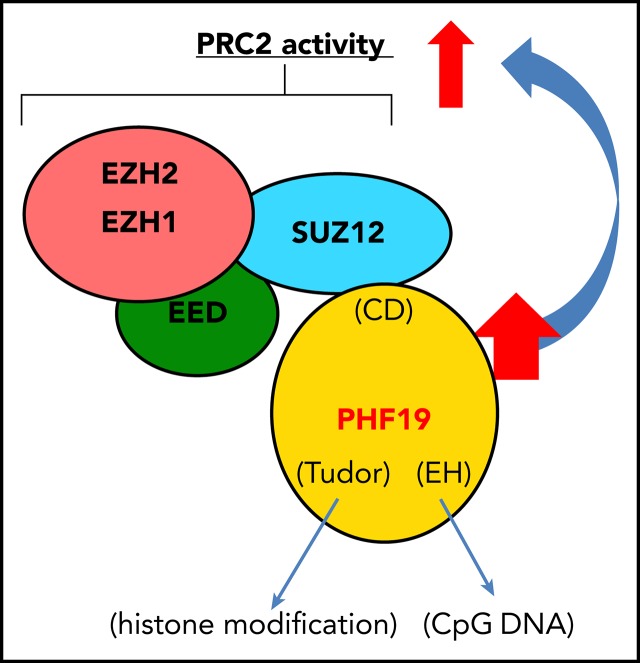
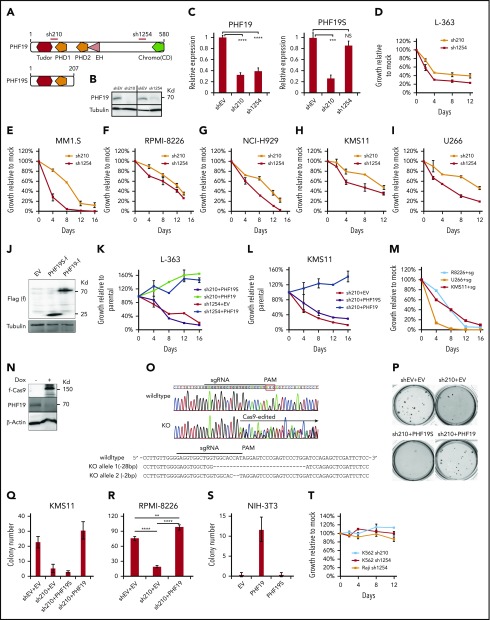
Dysregulation of polycomb repressive complex 2 (PRC2) promotes oncogenesis partly through its enzymatic function for inducing trimethylation of histone H3 lysine 27 (H3K27me3). However, it remains to be determined how PRC2 activity is regulated in normal and diseased settings. We here report a PRC2-associated cofactor, PHD finger protein 19 (PHF19; also known as polycomb-like 3), as a crucial mediator of tumorigenicity in multiple myeloma (MM). Overexpression and/or genomic amplification of PHF19 is found associated with malignant progression of MM and plasma cell leukemia, correlating to worse treatment outcomes. Using various MM models, we demonstrated a critical requirement of PHF19 for tumor growth in vitro and in vivo. Mechanistically, PHF19-mediated oncogenic effect relies on its PRC2-interacting and chromatin-binding functions. Chromatin immunoprecipitation followed by sequencing profiling showed a critical role for PHF19 in maintaining the H3K27me3 landscape. PHF19 depletion led to loss of broad H3K27me3 domains, possibly due to impaired H3K27me3 spreading from cytosine guanine dinucleotide islands, which is reminiscent to the reported effect of an "onco"-histone mutation, H3K27 to methionine (H3K27M). RNA-sequencing-based transcriptome profiling in MM lines also demonstrated a requirement of PHF19 for optimal silencing of PRC2 targets, which include cell cycle inhibitors and interferon-JAK-STAT signaling genes critically involved in tumor suppression. Correlation studies using patient sample data sets further support a clinical relevance of the PHF19-regulated pathways. Lastly, we show that MM cells are generally sensitive to PRC2 inhibitors. Collectively, this study demonstrates that PHF19 promotes MM tumorigenesis through enhancing H3K27me3 deposition and PRC2's gene-regulatory functions, lending support for PRC2 blockade as a means for MM therapeutics.
© 2019 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures






Comment in
-
Myeloma's sound of silencing.Blood. 2019 Oct 3;134(14):1116-1117. doi: 10.1182/blood.2019002742. Blood. 2019. PMID: 31582370 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA215284/CA/NCI NIH HHS/United States
- P01 CA196539/CA/NCI NIH HHS/United States
- UL1 TR003107/TR/NCATS NIH HHS/United States
- TL1 TR003109/TR/NCATS NIH HHS/United States
- R01 GM122749/GM/NIGMS NIH HHS/United States
- F31 CA232464/CA/NCI NIH HHS/United States
- P20 GM121293/GM/NIGMS NIH HHS/United States
- S10 OD018445/OD/NIH HHS/United States
- P20 GM103625/GM/NIGMS NIH HHS/United States
- T32 GM106999/GM/NIGMS NIH HHS/United States
- R01 CA211336/CA/NCI NIH HHS/United States
- R01 HL133120/HL/NHLBI NIH HHS/United States
- P30 CA016086/CA/NCI NIH HHS/United States
- P20 GM103429/GM/NIGMS NIH HHS/United States
- R01 AI118891/AI/NIAID NIH HHS/United States
- R01 GM110174/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
