

Dengue genetic divergence generates within-serotype antigenic variation, but serotypes dominate evolutionary dynamics
- PMID: 31385805
- PMCID: PMC6731059
- DOI: 10.7554/eLife.42496
Dengue genetic divergence generates within-serotype antigenic variation, but serotypes dominate evolutionary dynamics
Abstract
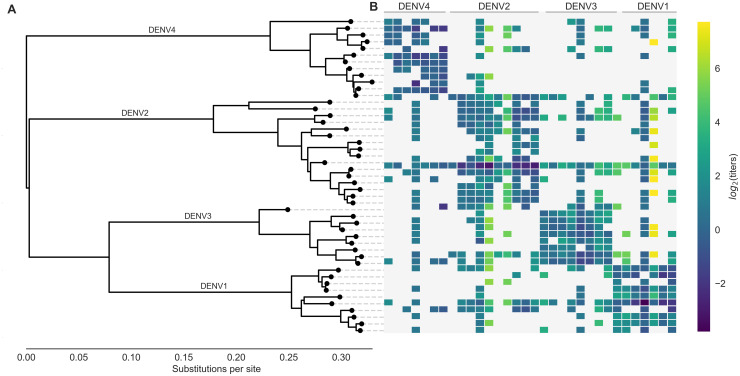
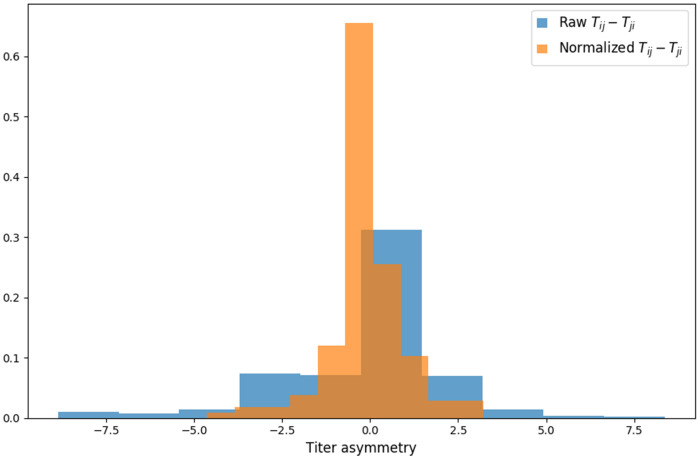
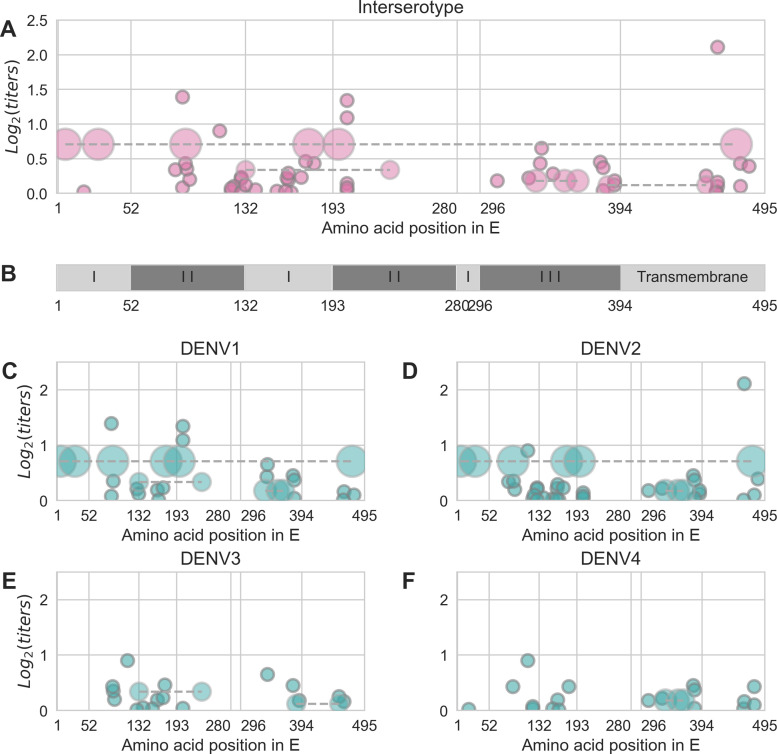
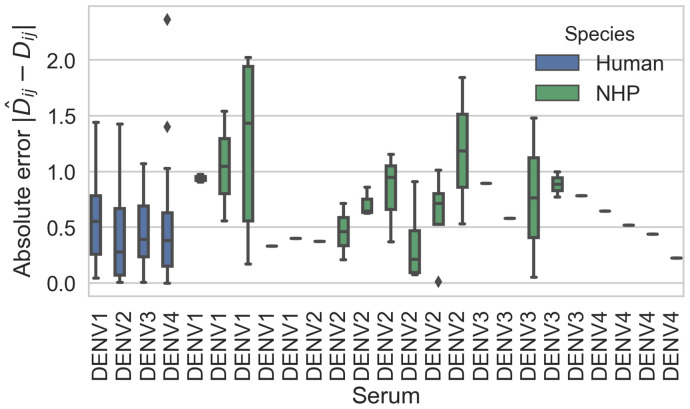
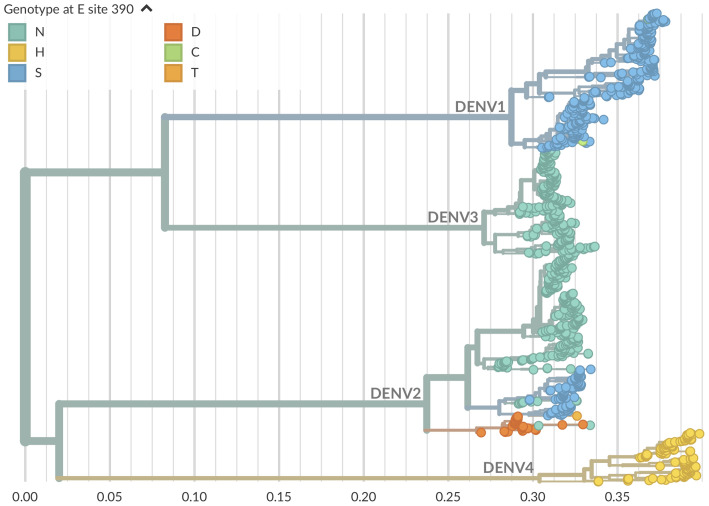
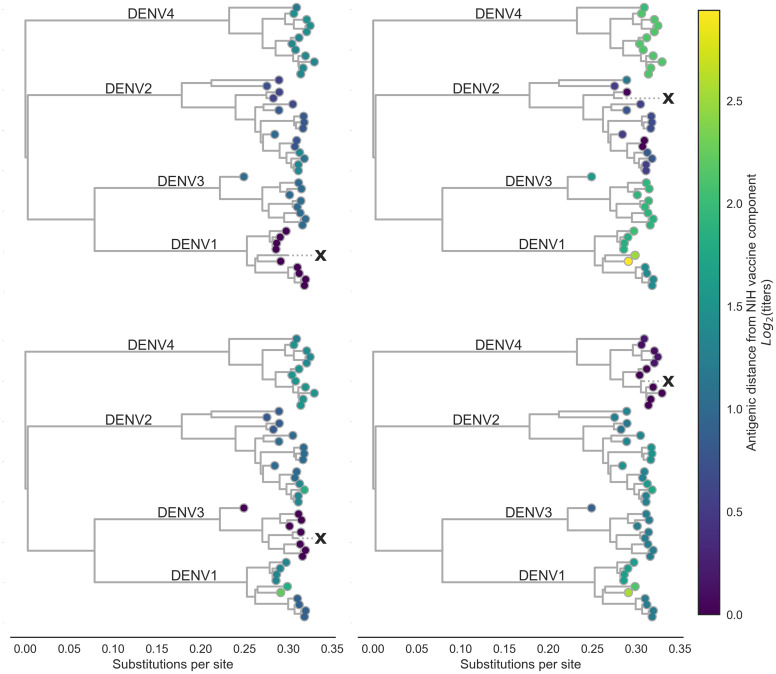
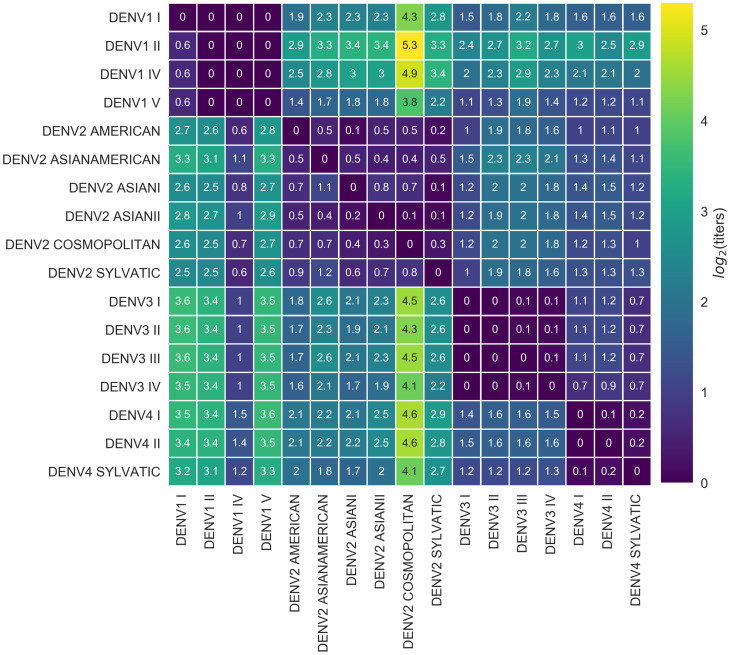
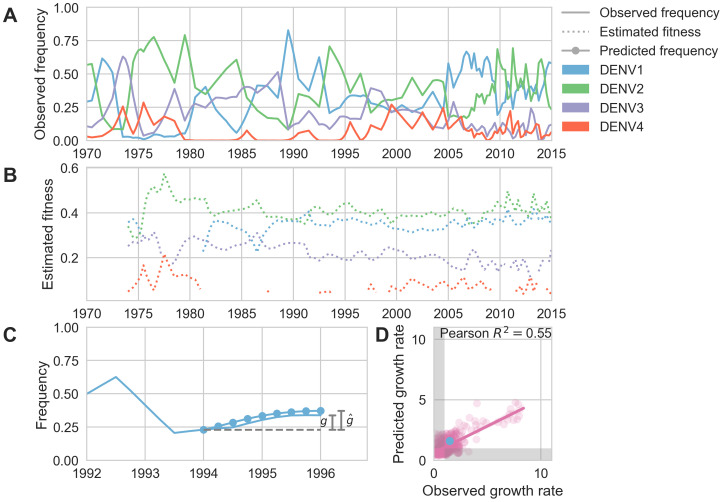
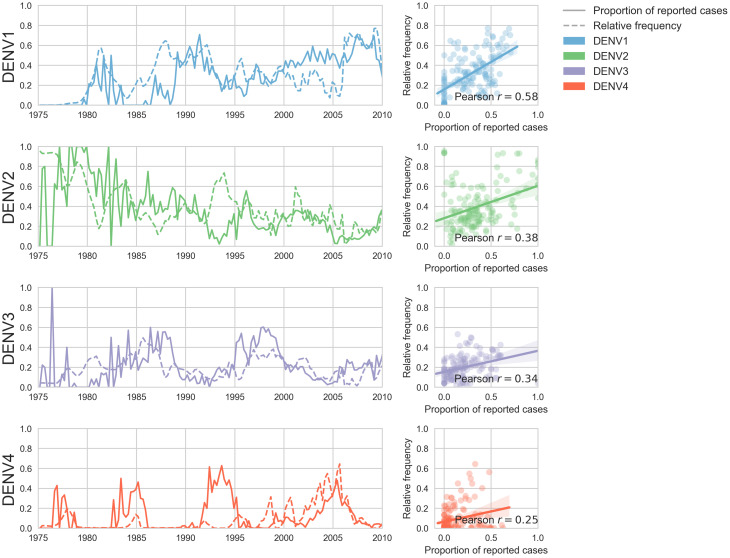
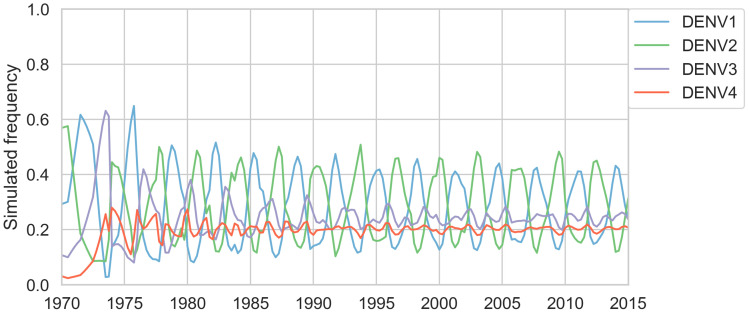
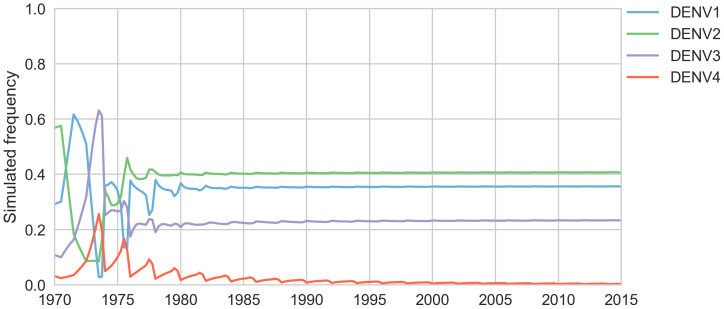
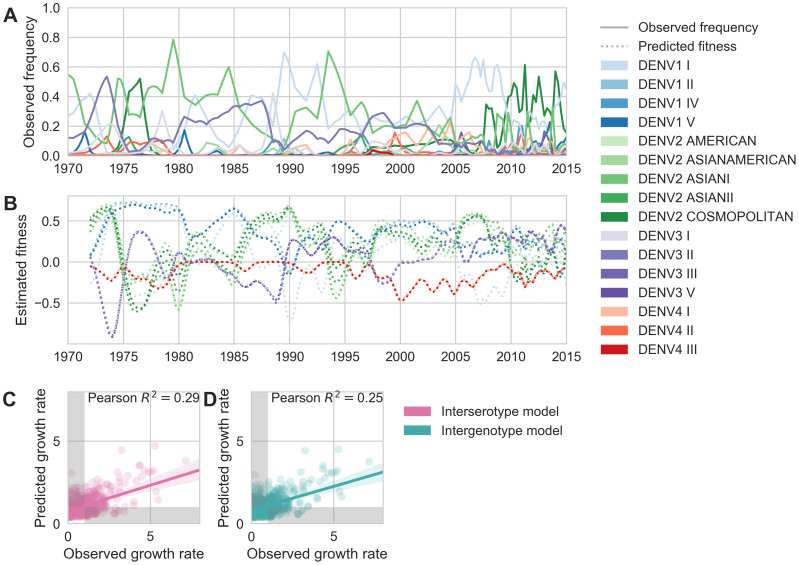
Dengue virus (DENV) exists as four genetically distinct serotypes, each of which is historically assumed to be antigenically uniform. Recent analyses suggest that antigenic heterogeneity may exist within each serotype, but its source, extent and impact remain unclear. Here, we construct a sequence-based model to directly map antigenic change to underlying genetic divergence. We identify 49 specific substitutions and four colinear substitution clusters that robustly predict dengue antigenic relationships. We report moderate antigenic diversity within each serotype, resulting in genotype-specific patterns of heterotypic cross-neutralization. We also quantify the impact of antigenic variation on real-world DENV population dynamics, and find that serotype-level antigenic fitness is a dominant driver of dengue clade turnover. These results provide a more nuanced understanding of the relationship between dengue genetic and antigenic evolution, and quantify the effect of antigenic fitness on dengue evolutionary dynamics.
Keywords: antigenic evolution; dengue; evolutionary biology; infectious disease; microbiology; viral fitness; virus.
© 2019, Bell et al.
Conflict of interest statement
SB, LK, TB No competing interests declared
Figures
References
-
- Andersen M, Dahl J, Vandenberghe L. CVXOPT: a Python package for convex optimization. Abel Ee Ucla Edu/cvxopt 2013
-
- Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. The global distribution and burden of dengue. Nature. 2013;496:504–507. doi: 10.1038/nature12060. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
