

STXBP1-associated neurodevelopmental disorder: a comparative study of behavioural characteristics
- PMID: 31387522
- PMCID: PMC6683428
- DOI: 10.1186/s11689-019-9278-9
STXBP1-associated neurodevelopmental disorder: a comparative study of behavioural characteristics
Abstract
Background: De novo loss of function mutations in STXBP1 are a relatively common cause of epilepsy and intellectual disability (ID). However, little is known about the types and severities of behavioural features associated with this genetic diagnosis.
Methods: To address this, we collected systematic phenotyping data encompassing neurological, developmental, and behavioural characteristics. Participants were 14 individuals with STXBP1-associated neurodevelopmental disorder, ascertained from clinical genetics and neurology services UK-wide. Data was collected via standardised questionnaires administered to parents at home, supplemented by researcher observations. To isolate discriminating phenotypes, the STXBP1 group was compared to 33 individuals with pathogenic mutations in other ID-associated genes (ID group). To account for the potential impact of global cognitive impairment, a secondary comparison was made to an ability-matched subset of the ID group (low-ability ID group).
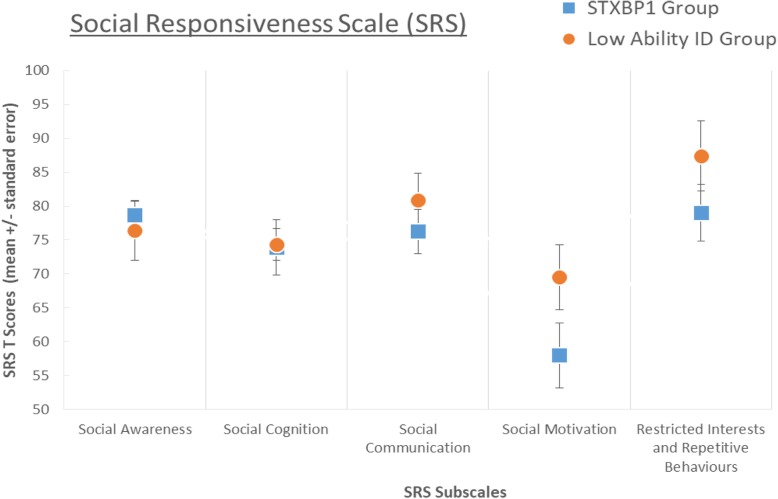
Results: The STXBP1 group demonstrated impairments across all assessed domains. In comparison to the ID group, the STXBP1 group had more severe global adaptive impairments, fine motor difficulties, and hyperactivity. In comparison to the low-ability ID group, severity of receptive language and social impairments discriminated the STXBP1 group. A striking feature of the STXBP1 group, with reference to both comparison groups, was preservation of social motivation.
Conclusions: De novo mutations in STXBP1 are associated with complex and variable neurodevelopmental impairments. Consistent features, which discriminate this disorder from other monogenic causes of ID, are severe language impairment and difficulties managing social interactions, despite strong social motivation. Future work could explore the physiological mechanisms linking motor, speech, and social development in this disorder. Understanding the developmental emergence of behavioural characteristics can help to focus clinical assessment and management after genetic diagnosis, with the long-term aim of improving outcomes for patients and families.
Keywords: Epilepsy; Intellectual disability; Language; STXBP1; Social.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Gilissen C, Hehir-Kwa JY, Thung DT, Van De Vorst M, Van Bon BWM, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511(7509):344–347. - PubMed
-
- Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40(6):782–788. - PubMed
-
- Deprez L, Weckhuysen S, Holmgren P, Suls A, Van Dyck T, Goossens D, et al. Clinical spectrum of early-onset epileptic encephalopathies associated with STXBP1 mutations. Neurology. 2010;75(13):1159–1165. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
