

Identification of Primary Antimicrobial Resistance Drivers in Agricultural Nontyphoidal Salmonella enterica Serovars by Using Machine Learning
- PMID: 31387929
- PMCID: PMC6687941
- DOI: 10.1128/mSystems.00211-19
Identification of Primary Antimicrobial Resistance Drivers in Agricultural Nontyphoidal Salmonella enterica Serovars by Using Machine Learning
Abstract
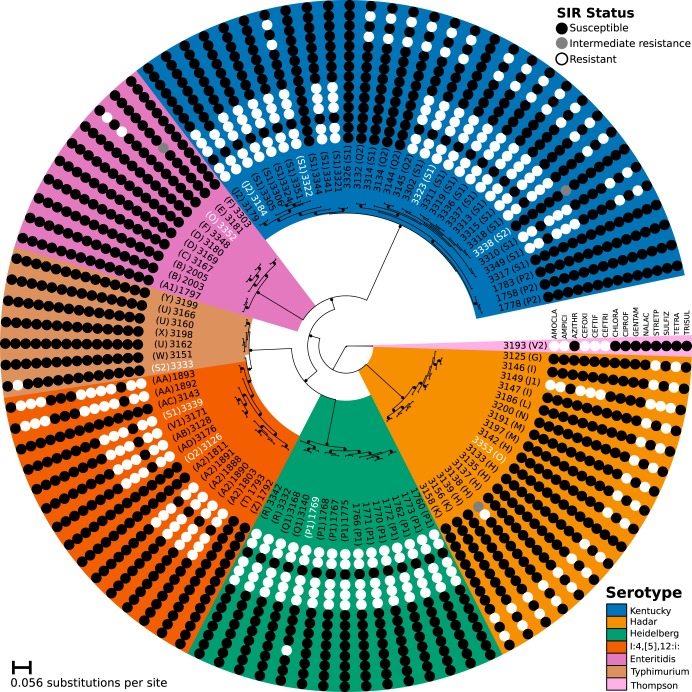
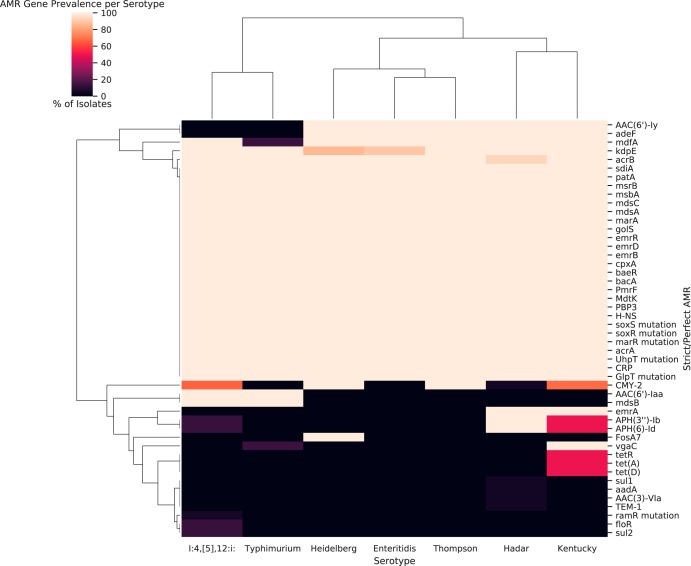
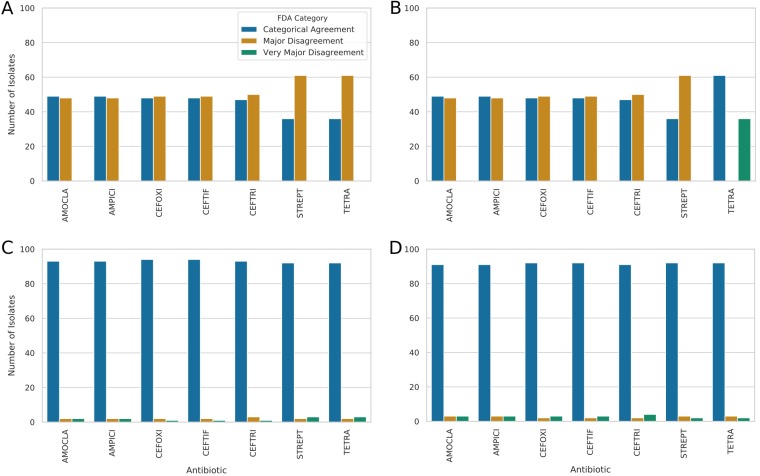
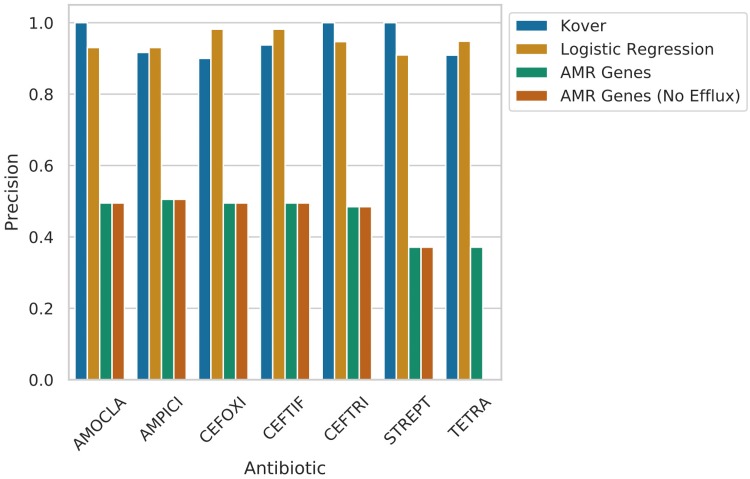
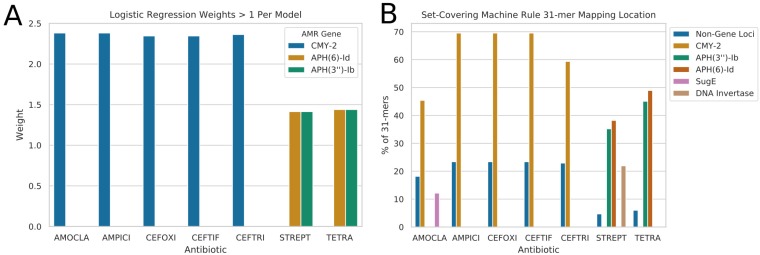
Nontyphoidal Salmonella (NTS) is a leading global cause of bacterial foodborne morbidity and mortality. Our ability to treat severe NTS infections has been impaired by increasing antimicrobial resistance (AMR). To understand and mitigate the global health crisis AMR represents, we need to link the observed resistance phenotypes with their underlying genomic mechanisms. Broiler chickens represent a key reservoir and vector for NTS infections, but isolates from this setting have been characterized in only very low numbers relative to clinical isolates. In this study, we sequenced and assembled 97 genomes encompassing 7 serotypes isolated from broiler chicken in farms in British Columbia between 2005 and 2008. Through application of machine learning (ML) models to predict the observed AMR phenotype from this genomic data, we were able to generate highly (0.92 to 0.99) precise logistic regression models using known AMR gene annotations as features for 7 antibiotics (amoxicillin-clavulanic acid, ampicillin, cefoxitin, ceftiofur, ceftriaxone, streptomycin, and tetracycline). Similarly, we also trained "reference-free" k-mer-based set-covering machine phenotypic prediction models (0.91 to 1.0 precision) for these antibiotics. By combining the inferred k-mers and logistic regression weights, we identified the primary drivers of AMR for the 7 studied antibiotics in these isolates. With our research representing one of the largest studies of a diverse set of NTS isolates from broiler chicken, we can thus confirm that the AmpC-like CMY-2 β-lactamase is a primary driver of β-lactam resistance and that the phosphotransferases APH(6)-Id and APH(3″-Ib) are the principal drivers of streptomycin resistance in this important ecosystem.IMPORTANCE Antimicrobial resistance (AMR) represents an existential threat to the function of modern medicine. Genomics and machine learning methods are being increasingly used to analyze and predict AMR. This type of surveillance is very important to try to reduce the impact of AMR. Machine learning models are typically trained using genomic data, but the aspects of the genomes that they use to make predictions are rarely analyzed. In this work, we showed how, by using different types of machine learning models and performing this analysis, it is possible to identify the key genes underlying AMR in nontyphoidal Salmonella (NTS). NTS is among the leading cause of foodborne illness globally; however, AMR in NTS has not been heavily studied within the food chain itself. Therefore, in this work we performed a broad-scale analysis of the AMR in NTS isolates from commercial chicken farms and identified some priority AMR genes for surveillance.
Keywords: AMR prediction; Salmonella; antimicrobial resistance; food chain; genomics; machine learning.
© Crown copyright 2019.
Figures
Similar articles
-
Genotypic Antimicrobial Resistance Profiles of Diarrheagenic Escherichia coli and Nontyphoidal Salmonella Strains Isolated from Children with Diarrhea and Their Exposure Environments in Ethiopia.Infect Drug Resist. 2024 Nov 9;17:4955-4972. doi: 10.2147/IDR.S480395. eCollection 2024. Infect Drug Resist. 2024. PMID: 39539744 Free PMC article.
-
Antimicrobial drug resistant non-typhoidal Salmonella enterica in commercial poultry value chain in Chitwan, Nepal.One Health Outlook. 2020 Oct 1;2:18. doi: 10.1186/s42522-020-00025-4. eCollection 2020. One Health Outlook. 2020. PMID: 33829137 Free PMC article.
-
Prevalence of antimicrobial resistance in fecal Escherichia coli and Salmonella enterica in Canadian commercial meat, companion, laboratory, and shelter rabbits (Oryctolagus cuniculus) and its association with routine antimicrobial use in commercial meat rabbits.Prev Vet Med. 2017 Nov 1;147:53-57. doi: 10.1016/j.prevetmed.2017.09.004. Epub 2017 Sep 6. Prev Vet Med. 2017. PMID: 29254727
-
Systematic Review and Meta-Analysis of Integrated Studies on Salmonella and Campylobacter Prevalence, Serovar, and Phenotyping and Genetic of Antimicrobial Resistance in the Middle East-A One Health Perspective.Antibiotics (Basel). 2022 Apr 19;11(5):536. doi: 10.3390/antibiotics11050536. Antibiotics (Basel). 2022. PMID: 35625181 Free PMC article. Review.
-
Antimicrobial Resistance in Nontyphoidal Salmonella Isolated from Human and Poultry-Related Samples in Brazil: 20-Year Meta-Analysis.Foodborne Pathog Dis. 2017 Feb;14(2):116-124. doi: 10.1089/fpd.2016.2228. Epub 2016 Dec 6. Foodborne Pathog Dis. 2017. PMID: 27922763
Cited by
-
An accurate and interpretable model for antimicrobial resistance in pathogenic Escherichia coli from livestock and companion animal species.PLoS One. 2023 Aug 24;18(8):e0290473. doi: 10.1371/journal.pone.0290473. eCollection 2023. PLoS One. 2023. PMID: 37616210 Free PMC article.
-
Identification of Novel Antimicrobial Resistance Genes Using Machine Learning, Homology Modeling, and Molecular Docking.Microorganisms. 2022 Oct 23;10(11):2102. doi: 10.3390/microorganisms10112102. Microorganisms. 2022. PMID: 36363694 Free PMC article.
-
Unique k-mers as Strain-Specific Barcodes for Phylogenetic Analysis and Natural Microbiome Profiling.Int J Mol Sci. 2020 Jan 31;21(3):944. doi: 10.3390/ijms21030944. Int J Mol Sci. 2020. PMID: 32023871 Free PMC article.
-
Predicting Salmonella MIC and Deciphering Genomic Determinants of Antibiotic Resistance and Susceptibility.Microorganisms. 2024 Jan 10;12(1):134. doi: 10.3390/microorganisms12010134. Microorganisms. 2024. PMID: 38257961 Free PMC article.
-
Machine learning and feature extraction for rapid antimicrobial resistance prediction of Acinetobacter baumannii from whole-genome sequencing data.Front Microbiol. 2024 Jan 11;14:1320312. doi: 10.3389/fmicb.2023.1320312. eCollection 2023. Front Microbiol. 2024. PMID: 38274740 Free PMC article.
References
-
- Robinson T, Bu D, Carrique-Mas J, Fèvre E, Gilbert M, Grace D, Hay S, Jiwakanon J, Kakkar M, Kariuki S, Laxminarayan R, Lubroth J, Magnusson U, Thi Ngoc P, van Boeckel TP, Woolhouse M. 2016. Antibiotic resistance is the quintessential One Health issue. Trans R Soc Trop Med Hyg 110:377–380. doi:10.1093/trstmh/trw048. - DOI - PMC - PubMed
-
- World Health Organization. 2015. Global action plan on antimicrobial resistance. World Health Organization, Geneva, Switzerland.
-
- Bradley P, Gordon NC, Walker TM, Dunn L, Heys S, Huang B, Earle S, Pankhurst LJ, Anson L, De Cesare M, Piazza P, Votintseva A, Golubchik T, Wilson D, Wyllie D, Diel R, Niemann S, Feuerriegel S, Kohl T, Ismail N, Omar S, Smith E, Buck D, McVean G, Walker A, Peto T, Crook D, Iqbal Z. 2015. Rapid antibiotic-resistance predictions from genome sequence data for Staphylococcus aureus and Mycobacterium tuberculosis. Nat Commun 6:10063. doi:10.1038/ncomms10063. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
