

FRET-based analysis of the cardiac troponin T linker region reveals the structural basis of the hypertrophic cardiomyopathy-causing Δ160E mutation
- PMID: 31387947
- PMCID: PMC6779437
- DOI: 10.1074/jbc.RA118.005098
FRET-based analysis of the cardiac troponin T linker region reveals the structural basis of the hypertrophic cardiomyopathy-causing Δ160E mutation
Abstract
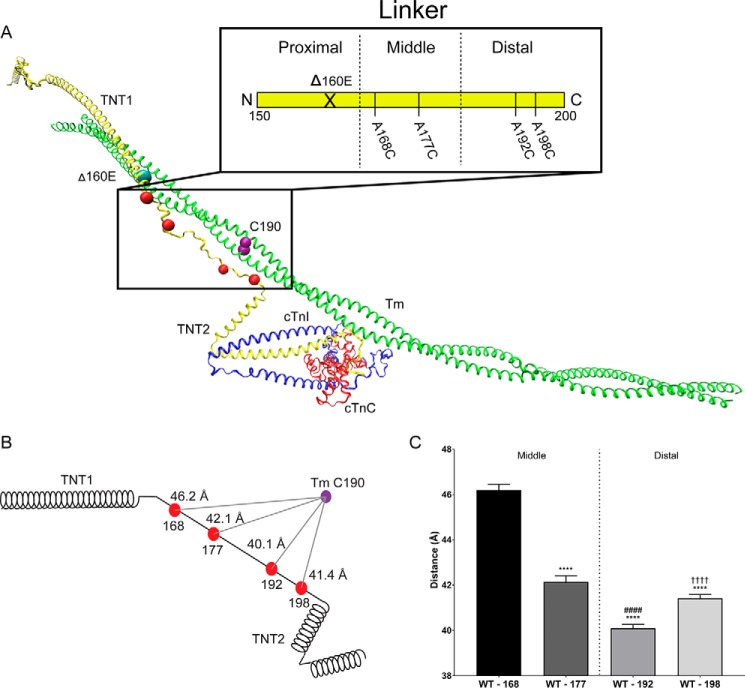
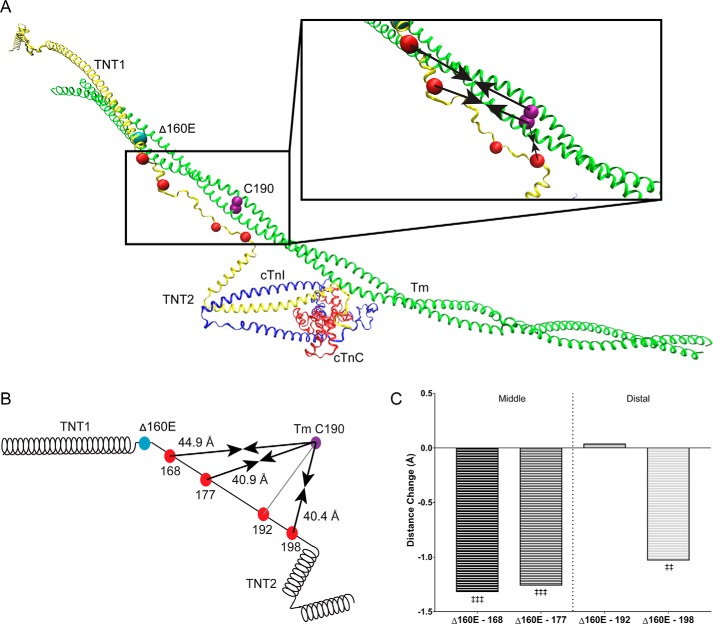
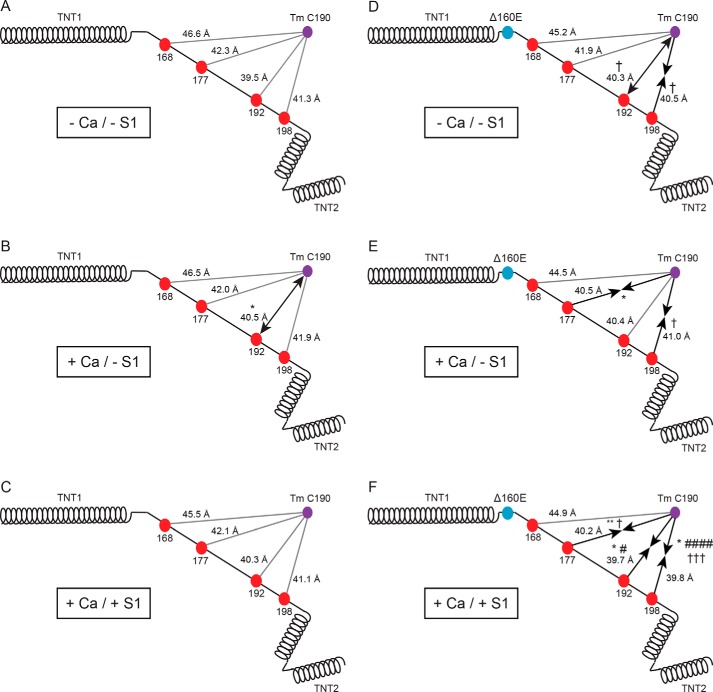
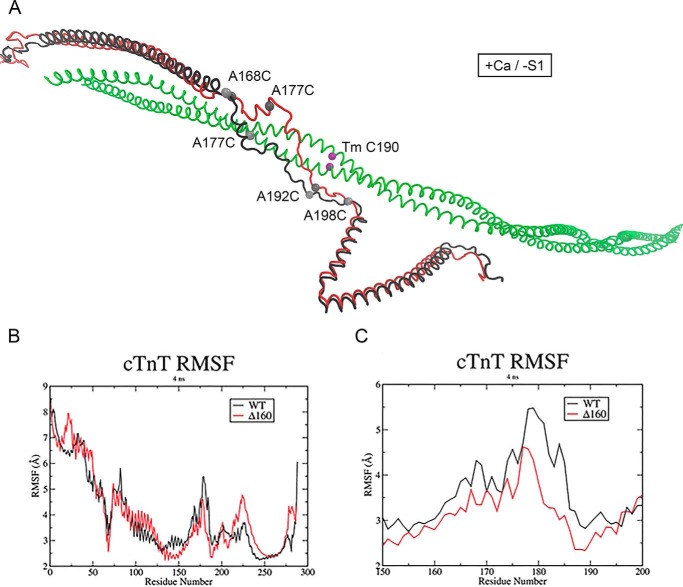
Mutations in the cardiac thin filament (TF) have highly variable effects on the regulatory function of the cardiac sarcomere. Understanding the molecular-level dysfunction elicited by TF mutations is crucial to elucidate cardiac disease mechanisms. The hypertrophic cardiomyopathy-causing cardiac troponin T (cTnT) mutation Δ160Glu (Δ160E) is located in a putative "hinge" adjacent to an unstructured linker connecting domains TNT1 and TNT2. Currently, no high-resolution structure exists for this region, limiting significantly our ability to understand its role in myofilament activation and the molecular mechanism of mutation-induced dysfunction. Previous regulated in vitro motility data have indicated mutation-induced impairment of weak actomyosin interactions. We hypothesized that cTnT-Δ160E repositions the flexible linker, altering weak actomyosin electrostatic binding and acting as a biophysical trigger for impaired contractility and the observed remodeling. Using time-resolved FRET and an all-atom TF model, here we first defined the WT structure of the cTnT-linker region and then identified Δ160E mutation-induced positional changes. Our results suggest that the WT linker runs alongside the C terminus of tropomyosin. The Δ160E-induced structural changes moved the linker closer to the tropomyosin C terminus, an effect that was more pronounced in the presence of myosin subfragment (S1) heads, supporting previous findings. Our in silico model fully supported this result, indicating a mutation-induced decrease in linker flexibility. Our findings provide a framework for understanding basic pathogenic mechanisms that drive severe clinical hypertrophic cardiomyopathy phenotypes and for identifying structural targets for intervention that can be tested in silico and in vitro.
Keywords: allosteric regulation; allostery; cardiac thin filament; cardiomyopathy; fluorescence resonance energy transfer (FRET); heart disease; hypertrophic cardiomyopathy; molecular dynamics; muscle contraction; mutant; troponin.
© 2019 Abdullah et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures
References
-
- Alcalai R., Seidman J. G., and Seidman C. E. (2008) Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J. Cardiovasc. Electrophysiol. 19, 104–110 - PubMed
-
- Kimura A. (2015) Molecular genetics and pathogenesis of cardiomyopathy. J. Hum. Genet. 61, 41–50 - PubMed
-
- Alfares A. A., Kelly M. A., McDermott G., Funke B. H., Lebo M. S., Baxter S. B., Shen J., McLaughlin H. M., Clark E. H., Babb L. J., Cox S. W., DePalma S. R., Ho C. Y., Seidman J. G., Seidman C. E., et al. (2015) Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet. Med. 17, 880–888 10.1038/gim.2014.205 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
