

The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis
- PMID: 31388190
- PMCID: PMC8900827
- DOI: 10.1038/s41436-019-0612-0
The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis
Erratum in
-
Correction: The CHD4-related syndrome: a comprehensive investigation of the clinical spectrum, genotype-phenotype correlations, and molecular basis.Genet Med. 2020 Mar;22(3):669. doi: 10.1038/s41436-019-0727-3. Genet Med. 2020. PMID: 31844176
Abstract
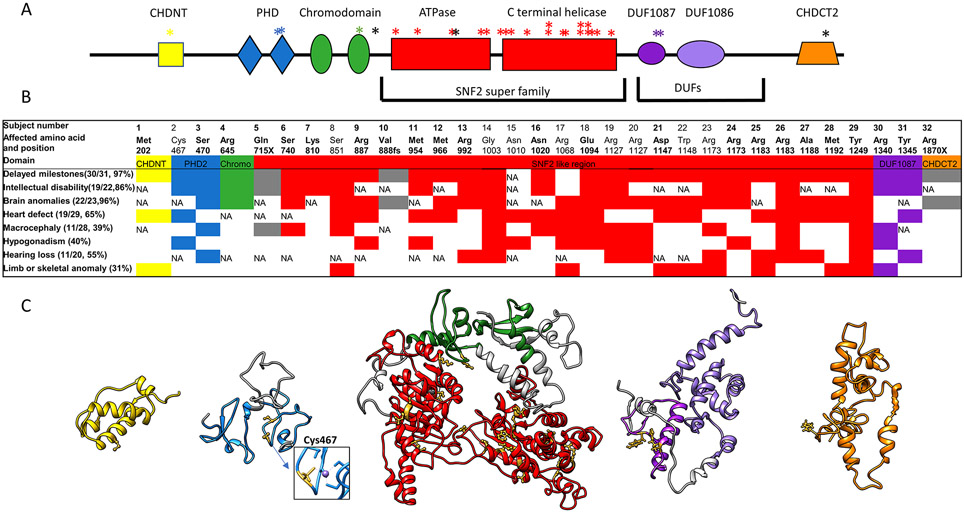
Purpose: Sifrim-Hitz-Weiss syndrome (SIHIWES) is a recently described multisystemic neurodevelopmental disorder caused by de novo variants inCHD4. In this study, we investigated the clinical spectrum of the disorder, genotype-phenotype correlations, and the effect of different missense variants on CHD4 function.
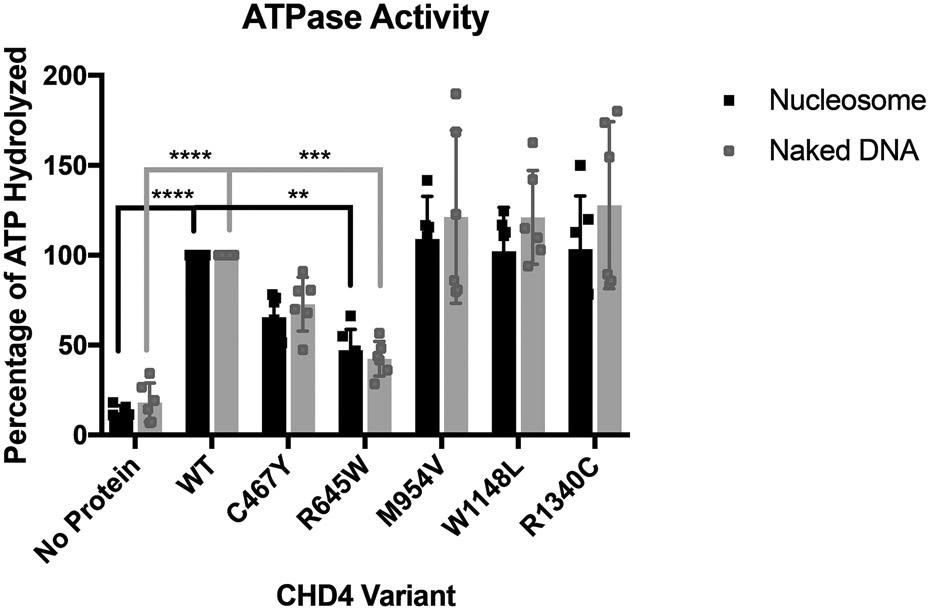
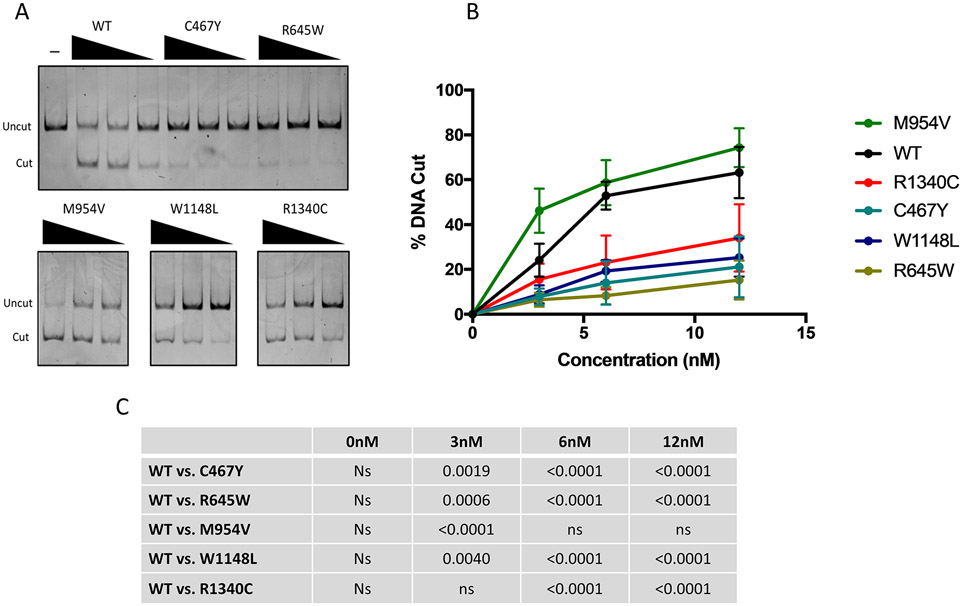
Methods: We collected clinical and molecular data from 32 individuals with mostly de novo variants in CHD4, identified through next-generation sequencing. We performed adenosine triphosphate (ATP) hydrolysis and nucleosome remodeling assays on variants from five different CHD4 domains.
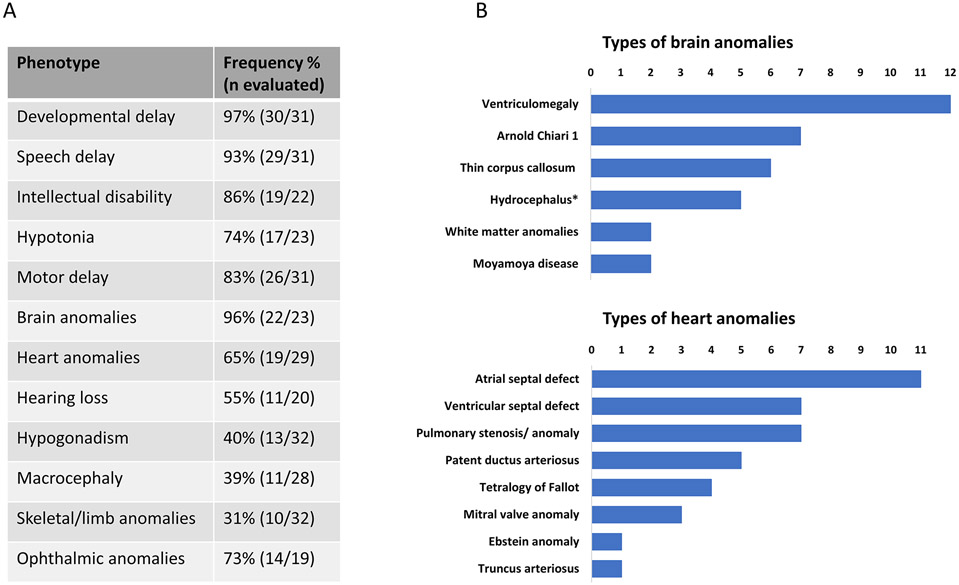
Results: The majority of participants had global developmental delay, mild to moderate intellectual disability, brain anomalies, congenital heart defects, and dysmorphic features. Macrocephaly was a frequent but not universal finding. Additional common abnormalities included hypogonadism in males, skeletal and limb anomalies, hearing impairment, and ophthalmic abnormalities. The majority of variants were nontruncating and affected the SNF2-like region of the protein. We did not identify genotype-phenotype correlations based on the type or location of variants. Alterations in ATP hydrolysis and chromatin remodeling activities were observed in variants from different domains.
Conclusion: The CHD4-related syndrome is a multisystemic neurodevelopmental disorder. Missense substitutions in different protein domains alter CHD4 function in a variant-specific manner, but result in a similar phenotype in humans.
Keywords: 12p13.31; ATPase; chromatin remodeling; intellectual disability; missense.
Figures

References
-
- Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2(6):851–861. http://www.ncbi.nlm.nih.gov/pubmed/9885572. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UM1 HL098162/HL/NHLBI NIH HHS/United States
- UM1 HL098123/HL/NHLBI NIH HHS/United States
- U01 HL131003/HL/NHLBI NIH HHS/United States
- S10 OD026880/OD/NIH HHS/United States
- HICF-1009-003/DH_/Department of Health/United Kingdom
- UM1 HL098147/HL/NHLBI NIH HHS/United States
- WT098051/WT_/Wellcome Trust/United Kingdom
- UM1 HL128761/HL/NHLBI NIH HHS/United States
- ZIA ES101965/ImNIH/Intramural NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- UM1 HL128711/HL/NHLBI NIH HHS/United States
- Z01 ES101965/ImNIH/Intramural NIH HHS/United States
- UM1 HL172717/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
