

MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies
- PMID: 31388474
- PMCID: PMC6662567
- DOI: 10.7717/peerj.7359
MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies
Abstract
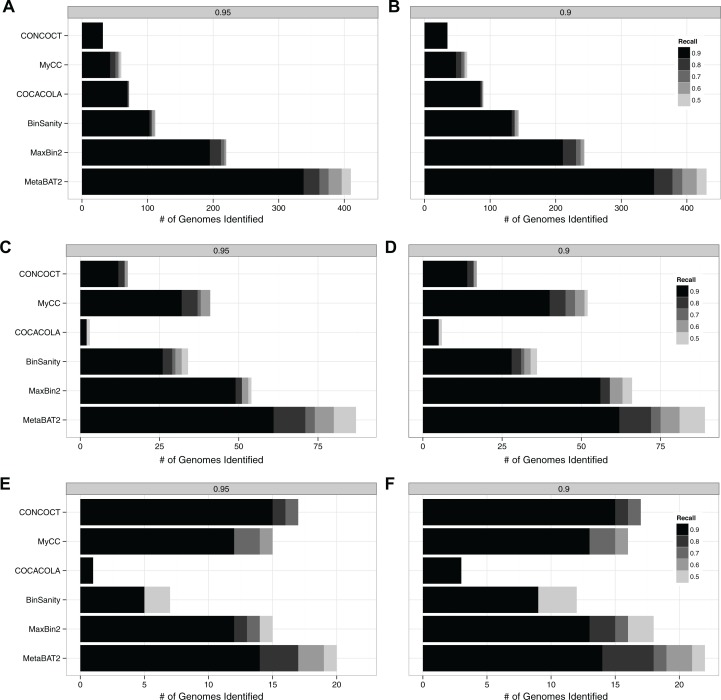
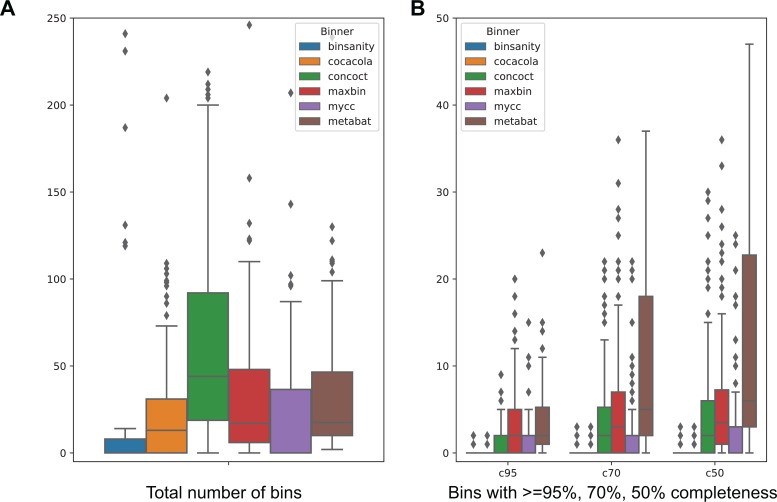
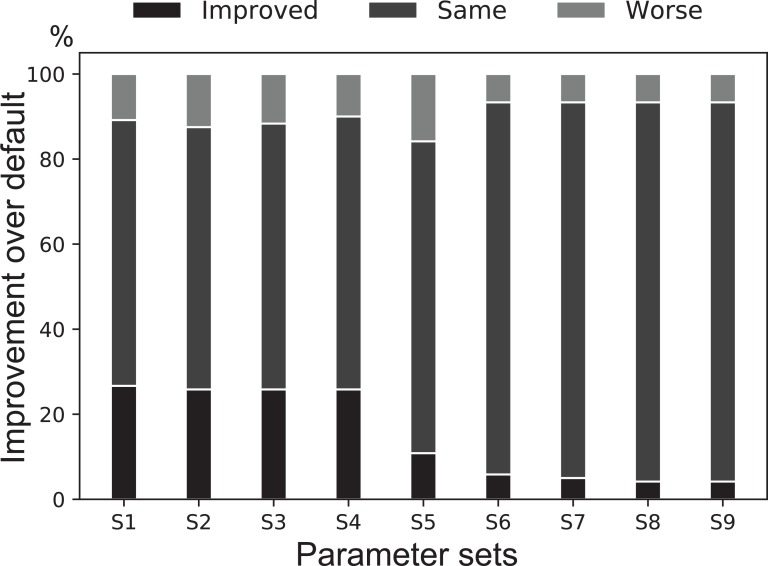
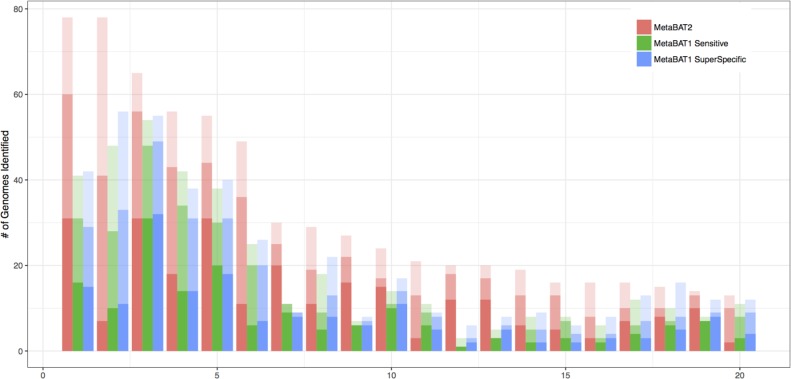
We previously reported on MetaBAT, an automated metagenome binning software tool to reconstruct single genomes from microbial communities for subsequent analyses of uncultivated microbial species. MetaBAT has become one of the most popular binning tools largely due to its computational efficiency and ease of use, especially in binning experiments with a large number of samples and a large assembly. MetaBAT requires users to choose parameters to fine-tune its sensitivity and specificity. If those parameters are not chosen properly, binning accuracy can suffer, especially on assemblies of poor quality. Here, we developed MetaBAT 2 to overcome this problem. MetaBAT 2 uses a new adaptive binning algorithm to eliminate manual parameter tuning. We also performed extensive software engineering optimization to increase both computational and memory efficiency. Comparing MetaBAT 2 to alternative software tools on over 100 real world metagenome assemblies shows superior accuracy and computing speed. Binning a typical metagenome assembly takes only a few minutes on a single commodity workstation. We therefore recommend the community adopts MetaBAT 2 for their metagenome binning experiments. MetaBAT 2 is open source software and available at https://bitbucket.org/berkeleylab/metabat.
Keywords: Clustering; Metagenome binning; Metagenomics.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Bahram M, Hildebrand F, Forslund SK, Anderson JL, Soudzilovskaia NA, Bodegom PM, Bengtsson-Palme J, Anslan S, Coelho LP, Harend H, Huerta-Cepas J, Medema MH, Maltz MR, Mundra S, Olsson PA, Pent M, Põlme S, Sunagawa S, Ryberg M, Tedersoo L, Bork P. Structure and function of the global topsoil microbiome. Nature. 2018;560(7717):233–237. doi: 10.1038/s41586-018-0386-6. - DOI - PubMed
-
- Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu W-T, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Lapidus A, Meyer F, Yilmaz P, Parks DH, Eren AM, Schriml L, Banfield JF, Hugenholtz P, Woyke T, Genome Standards Consortium Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nature Biotechnology. 2017;35(8):725–731. doi: 10.1038/nbt.3893. - DOI - PMC - PubMed
-
- Chen I-MA, Chu K, Palaniappan K, Pillay M, Ratner A, Huang J, Huntemann M, Varghese N, White JR, Seshadri R, Smirnova T, Kirton E, Jungbluth SP, Woyke T, Eloe-Fadrosh EA, Ivanova NN, Kyrpides NC. IMG/M v. 5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Research. 2018;47(D1):D666–D677. - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
