

Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells
- PMID: 31391321
- PMCID: PMC7568232
- DOI: 10.1126/scitranslmed.aau4972
Targeting pyrimidine synthesis accentuates molecular therapy response in glioblastoma stem cells
Abstract
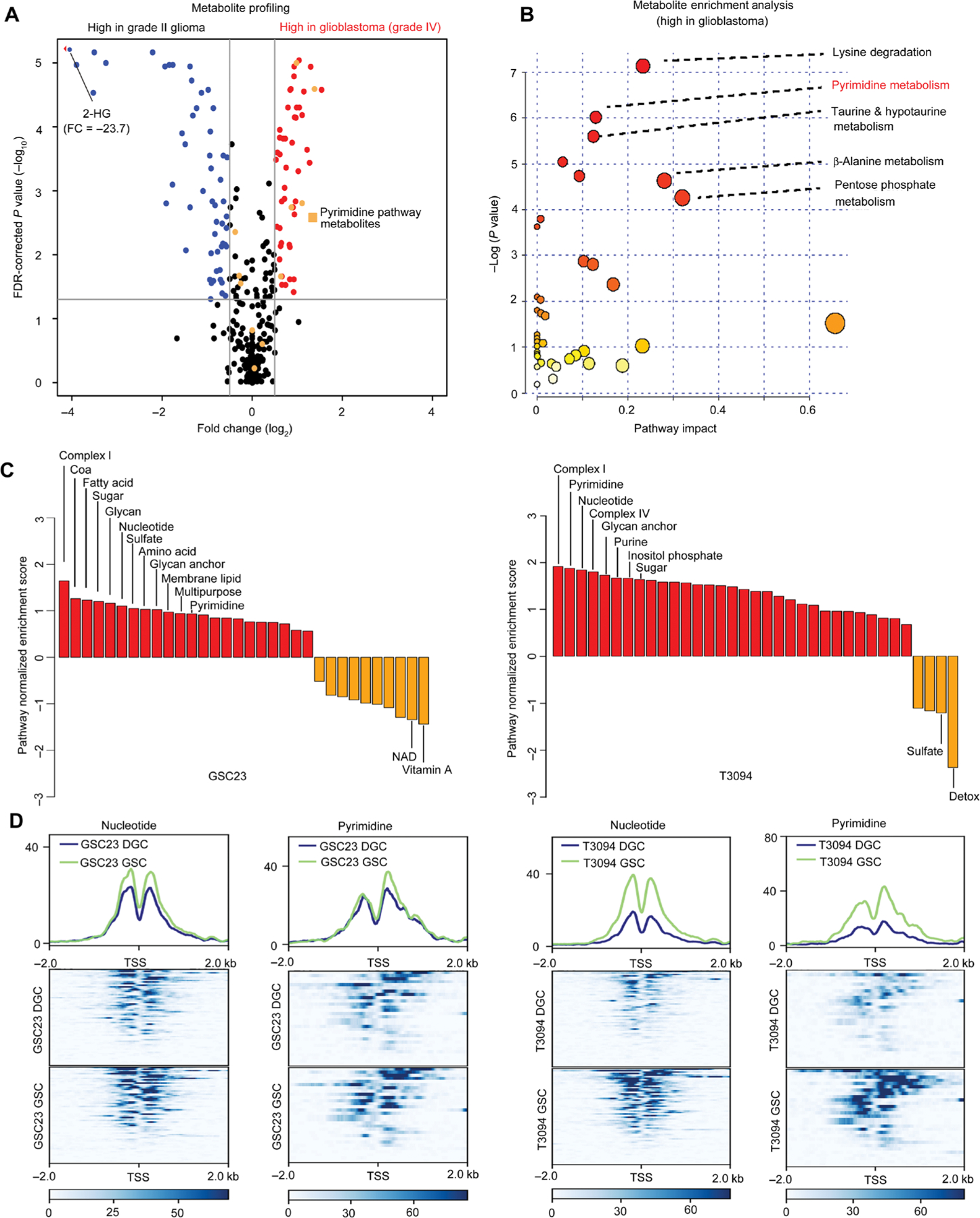
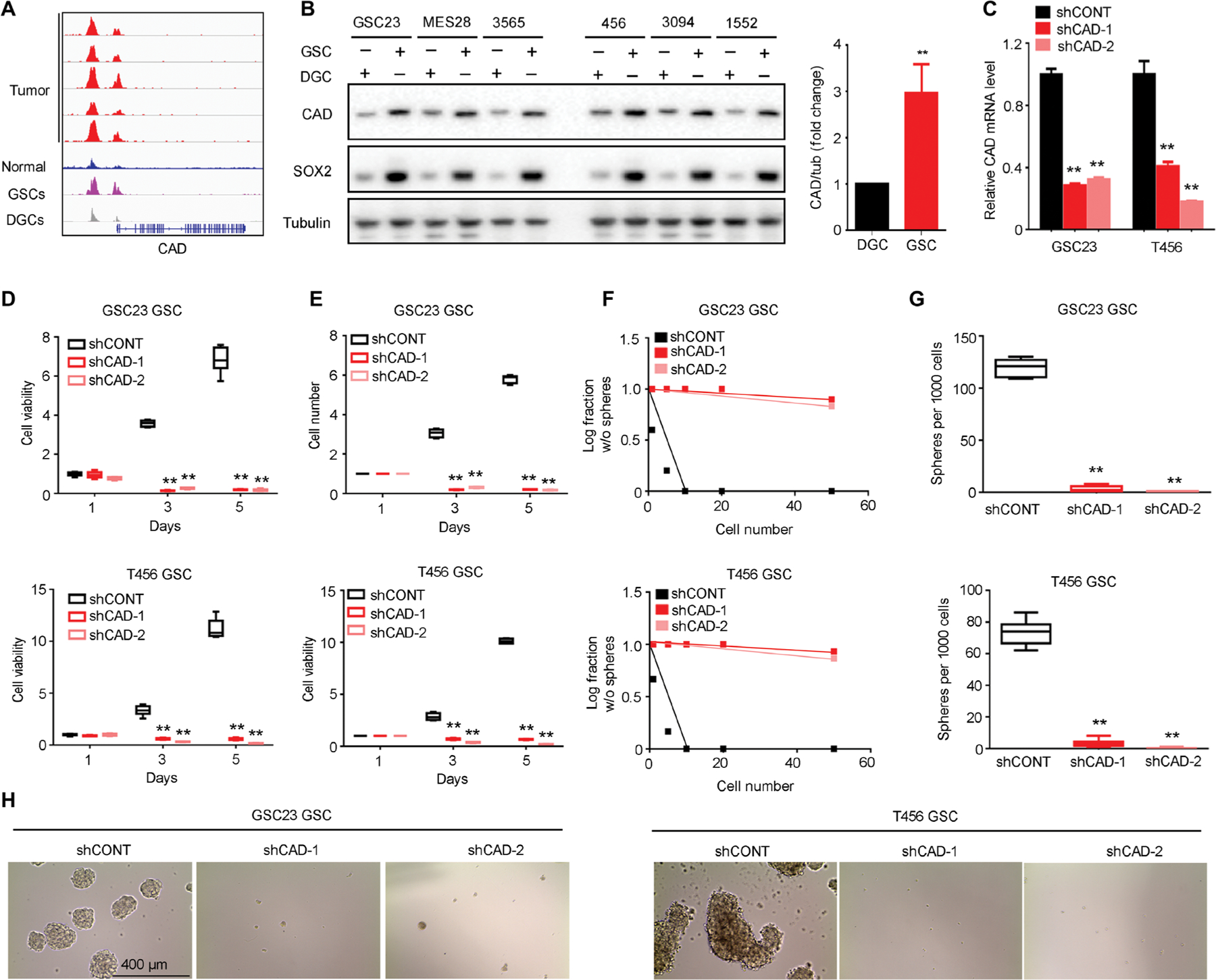
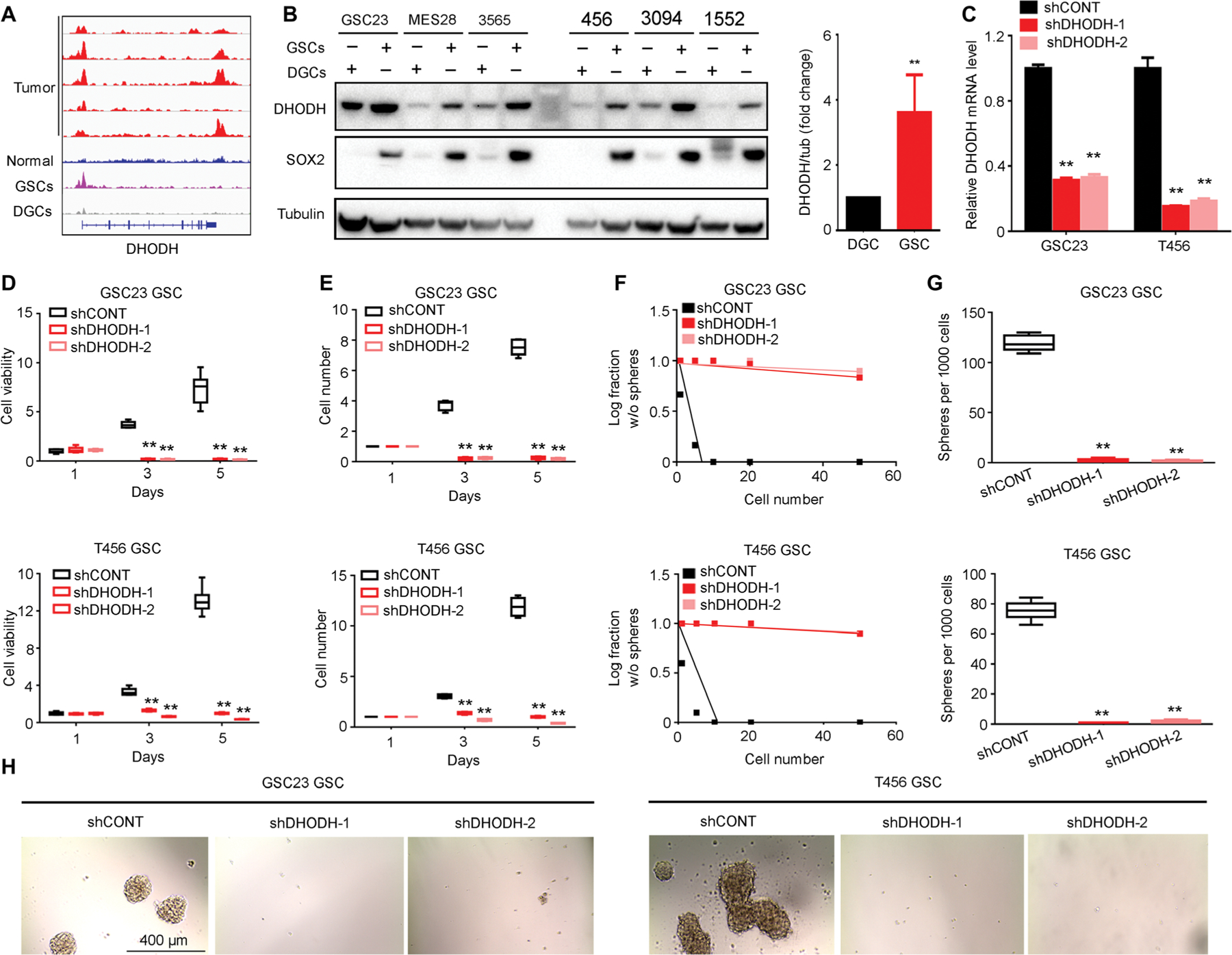
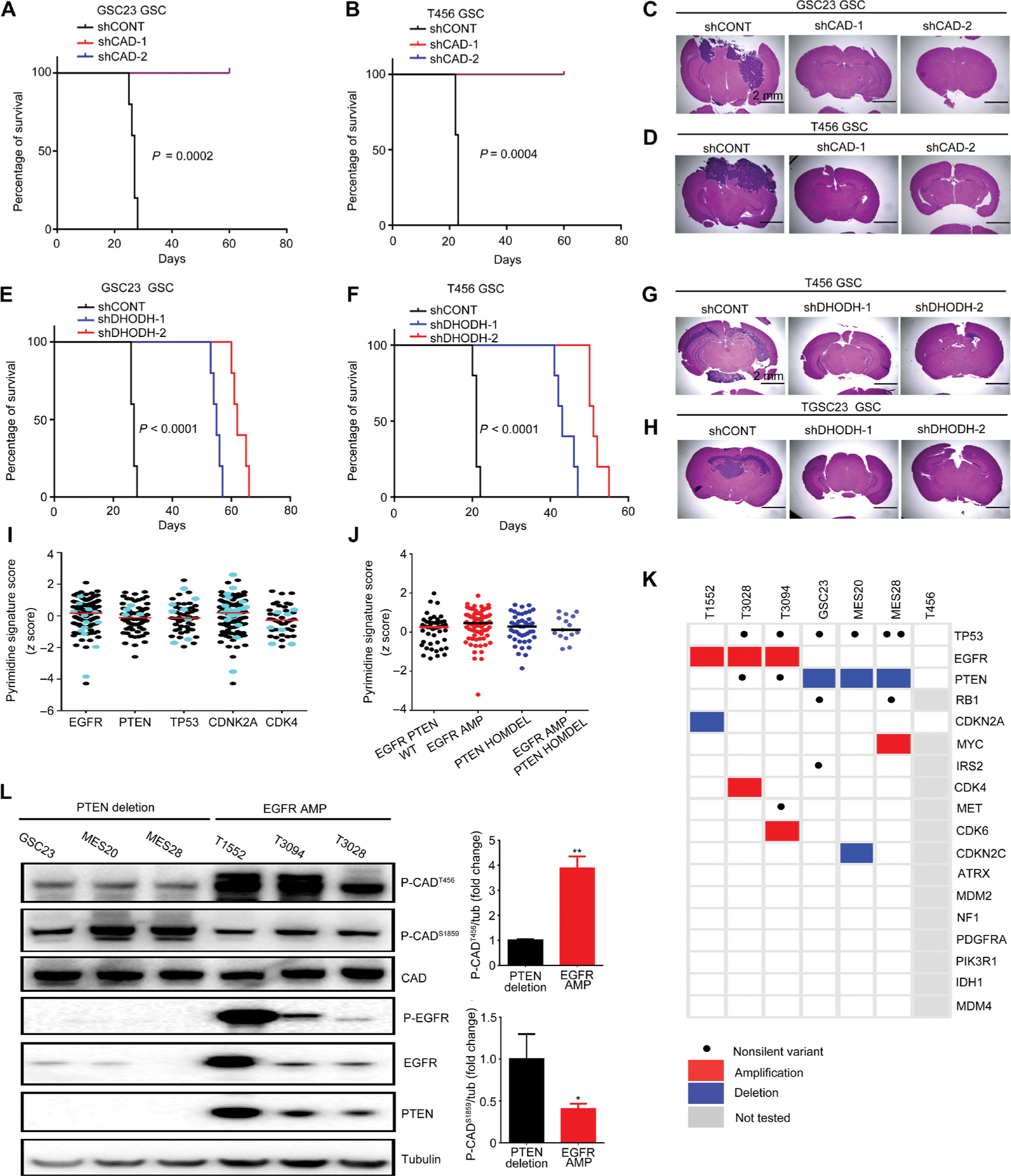
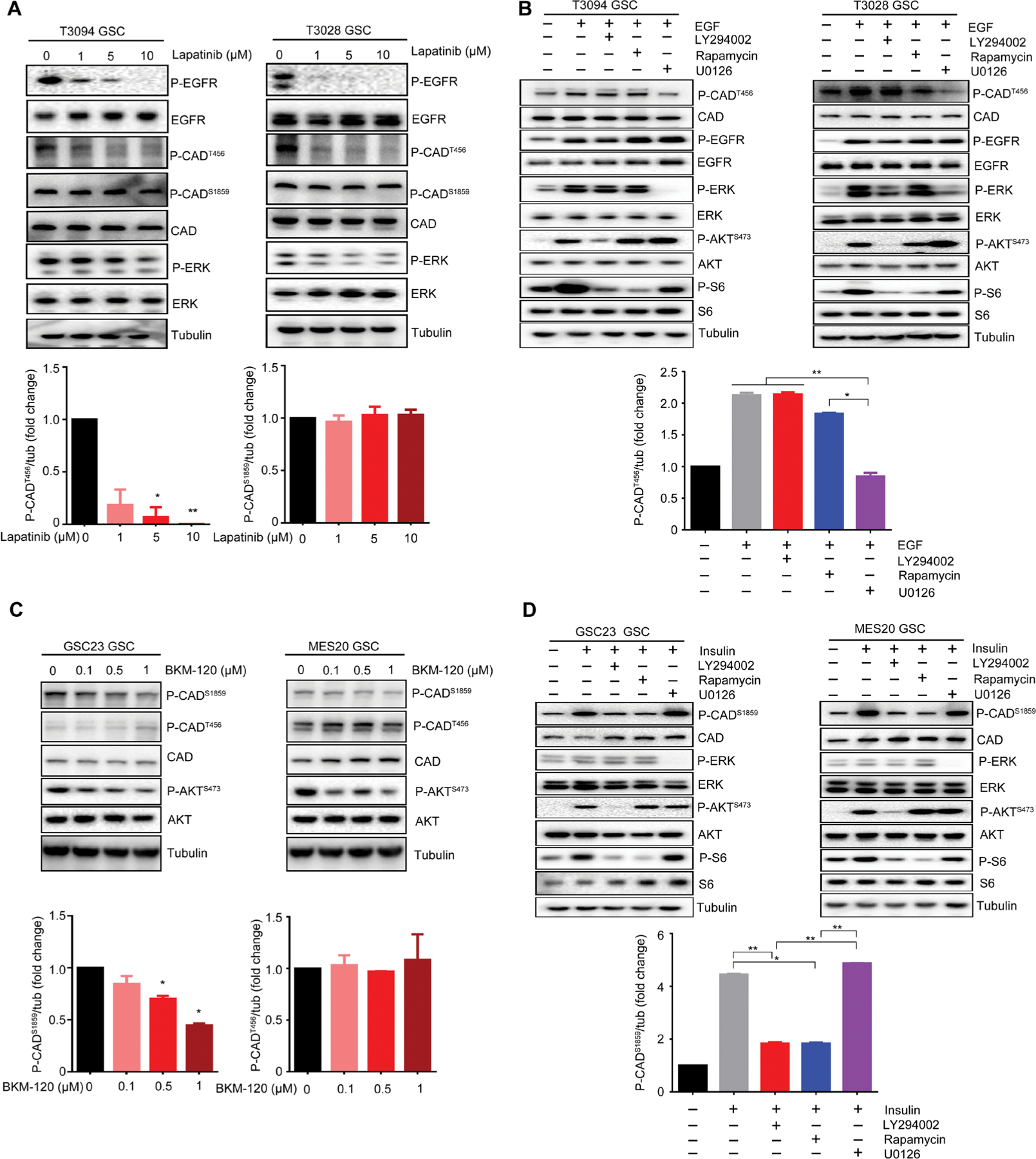
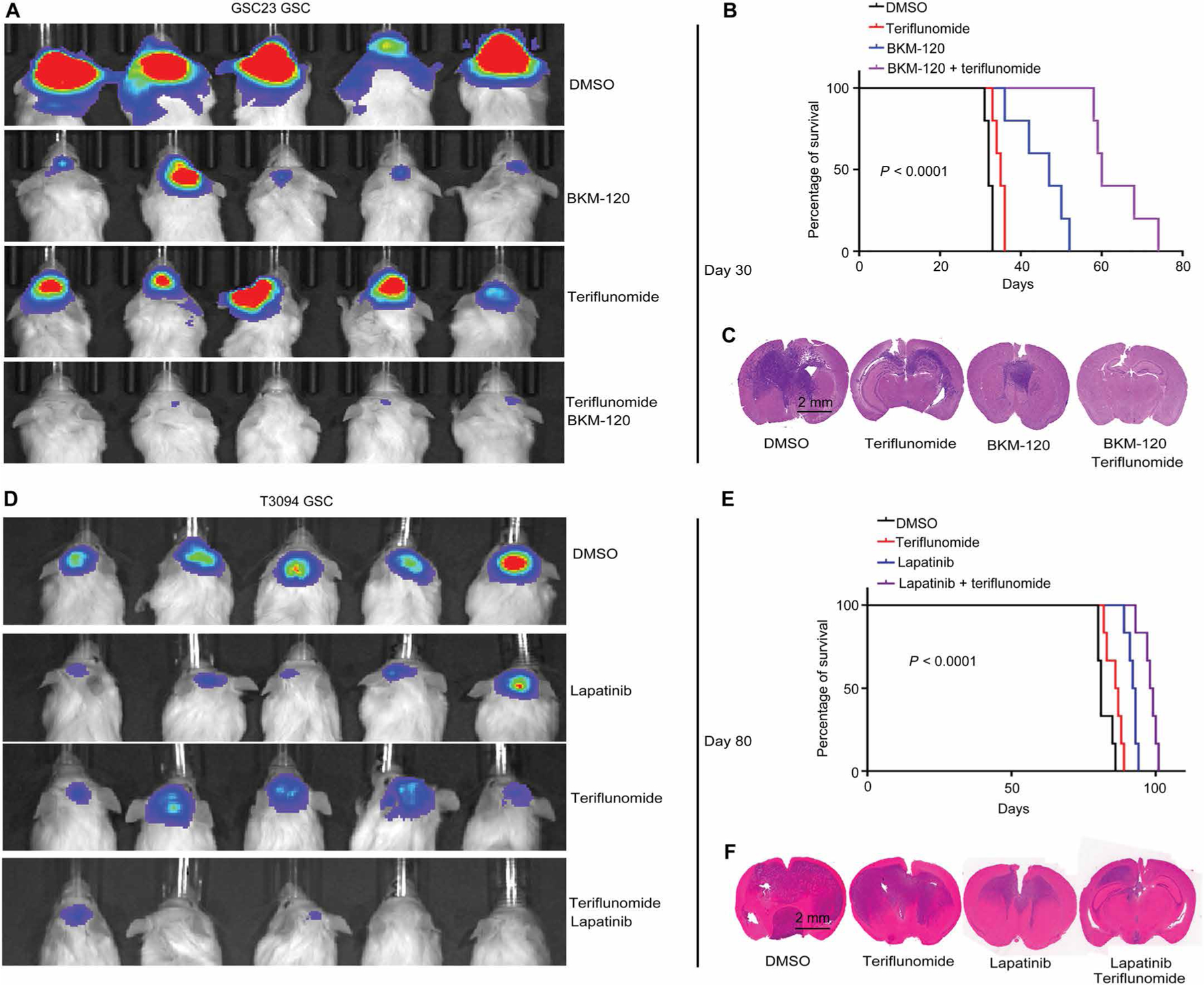
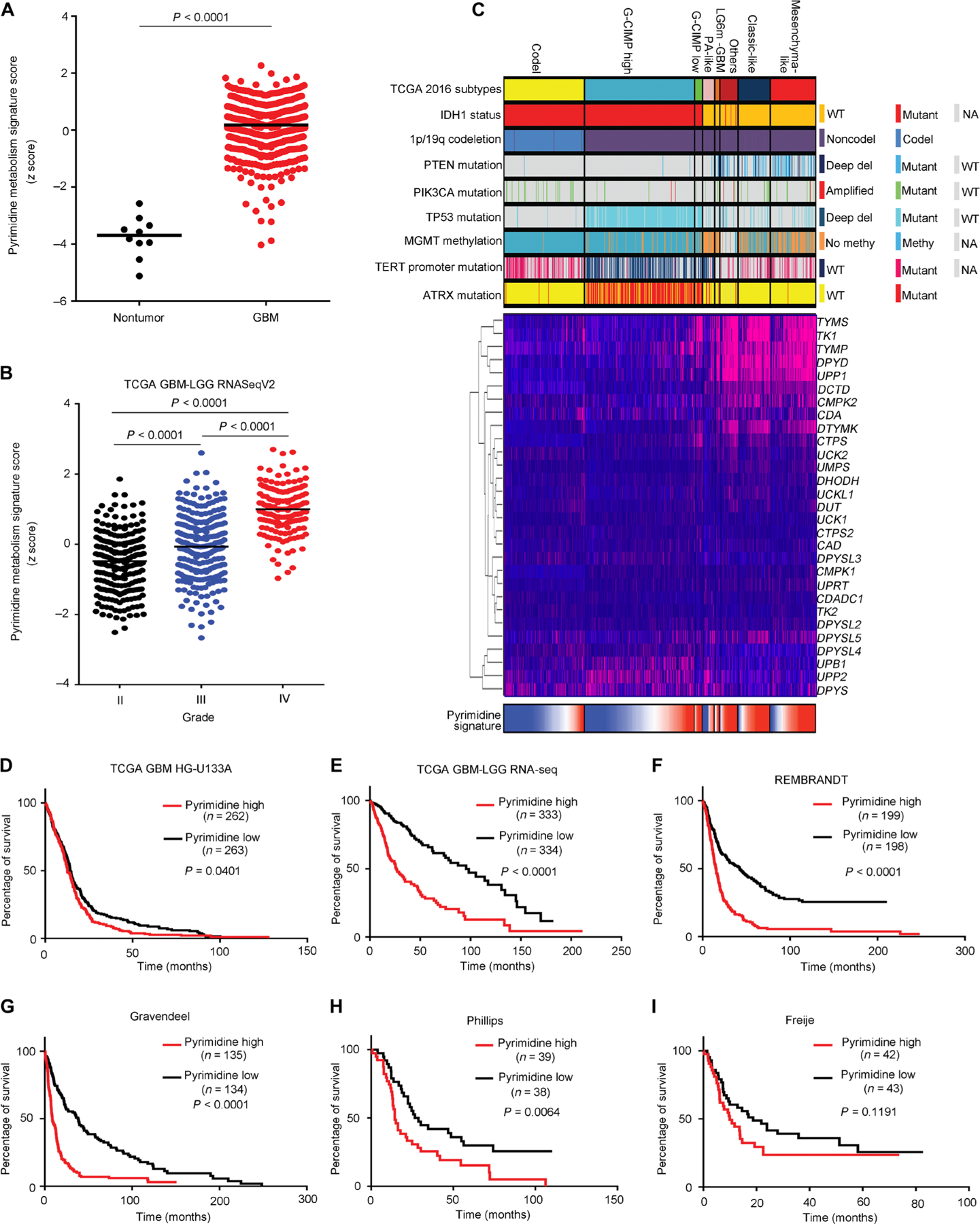
Glioblastoma stem cells (GSCs) reprogram glucose metabolism by hijacking high-affinity glucose uptake to survive in a nutritionally dynamic microenvironment. Here, we trace metabolic aberrations in GSCs to link core genetic mutations in glioblastoma to dependency on de novo pyrimidine synthesis. Targeting the pyrimidine synthetic rate-limiting step enzyme carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, dihydroorotase (CAD) or the critical downstream enzyme dihydroorotate dehydrogenase (DHODH) inhibited GSC survival, self-renewal, and in vivo tumor initiation through the depletion of the pyrimidine nucleotide supply in rodent models. Mutations in EGFR or PTEN generated distinct CAD phosphorylation patterns to activate carbon influx through pyrimidine synthesis. Simultaneous abrogation of tumor-specific driver mutations and DHODH activity with clinically approved inhibitors demonstrated sustained inhibition of metabolic activity of pyrimidine synthesis and GSC tumorigenic capacity in vitro. Higher expression of pyrimidine synthesis genes portends poor prognosis of patients with glioblastoma. Collectively, our results demonstrate a therapeutic approach of precision medicine through targeting the nexus between driver mutations and metabolic reprogramming in cancer stem cells.
Copyright © 2019 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Figures
References
-
- Ostrom QT, Gittleman H, de Blank PM, Finlay JL, Gurney JG, McKean-Cowdin R, Stearns DS, Wolff JE, Liu M, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS, American Brain Tumor Association adolescent and young adult primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 18 (suppl. 1), i1–i50 (2016). - PMC - PubMed
-
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO; European Organisation for Research and Treatment of Cancer Brain, Tumuour amd Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials, Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009). - PubMed
-
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, Louis DN, Rozenblatt-Rosen O, Suva ML, Regev A, Bernstein BE, Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401 (2014). - PMC - PubMed
-
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN, Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756–760 (2006). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA169117/CA/NCI NIH HHS/United States
- R01 NS073831/NS/NINDS NIH HHS/United States
- R01 CA129958/CA/NCI NIH HHS/United States
- R01 NS087913/NS/NINDS NIH HHS/United States
- R01 NS103434/NS/NINDS NIH HHS/United States
- R35 CA197718/CA/NCI NIH HHS/United States
- R01 NS091080/NS/NINDS NIH HHS/United States
- R01 CA184090/CA/NCI NIH HHS/United States
- TL1 TR002549/TR/NCATS NIH HHS/United States
- R01 CA116659/CA/NCI NIH HHS/United States
- R01 NS099175/NS/NINDS NIH HHS/United States
- R01 CA238662/CA/NCI NIH HHS/United States
- R01 CA171652/CA/NCI NIH HHS/United States
- R01 NS089272/NS/NINDS NIH HHS/United States
- T32 GM007250/GM/NIGMS NIH HHS/United States
- R01 CA154130/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
