

DISSEQT-DIStribution-based modeling of SEQuence space Time dynamics
- PMID: 31392032
- PMCID: PMC6680062
- DOI: 10.1093/ve/vez028
DISSEQT-DIStribution-based modeling of SEQuence space Time dynamics
Erratum in
-
Erratum: Santa Fe Institute Workshop Special Issue articles.Virus Evol. 2019 Nov 19;5(2):vez052. doi: 10.1093/ve/vez052. eCollection 2019 Jul. Virus Evol. 2019. PMID: 31768266 Free PMC article.
Abstract
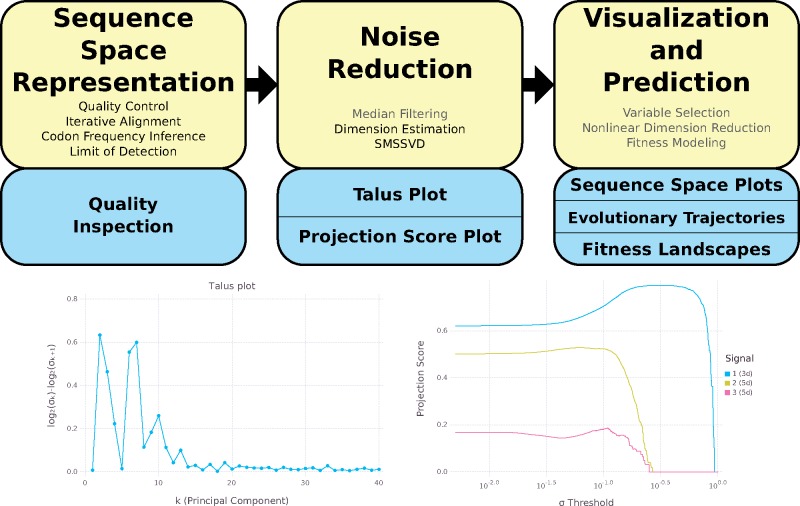
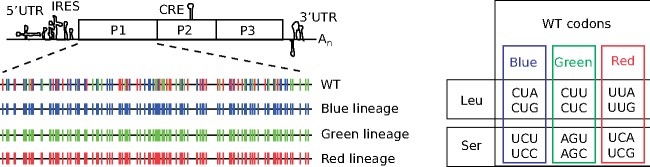
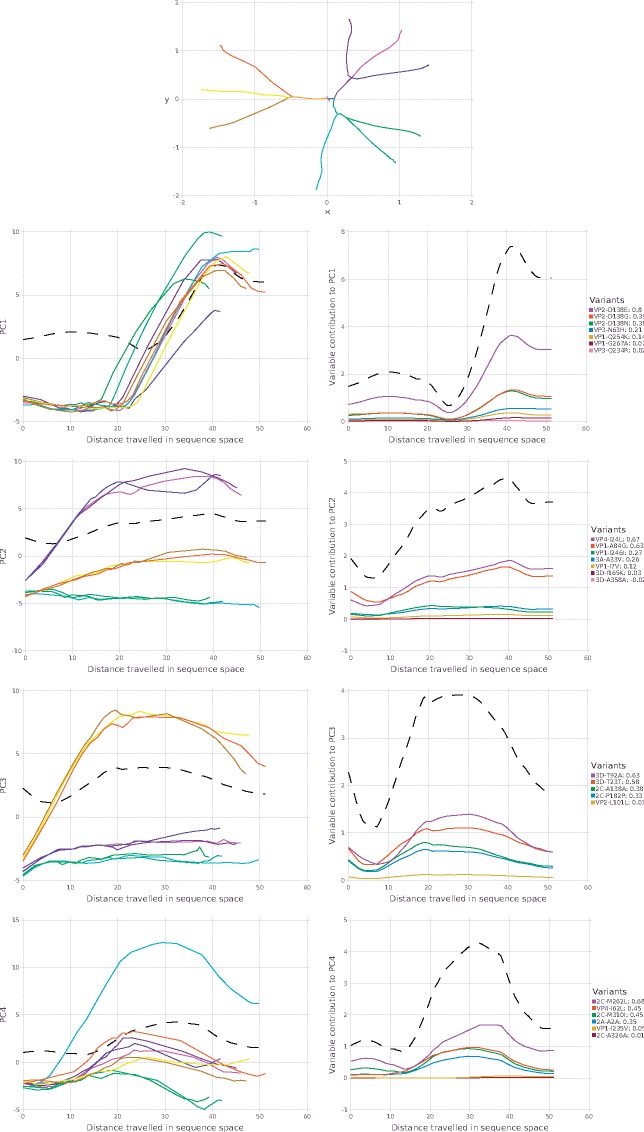
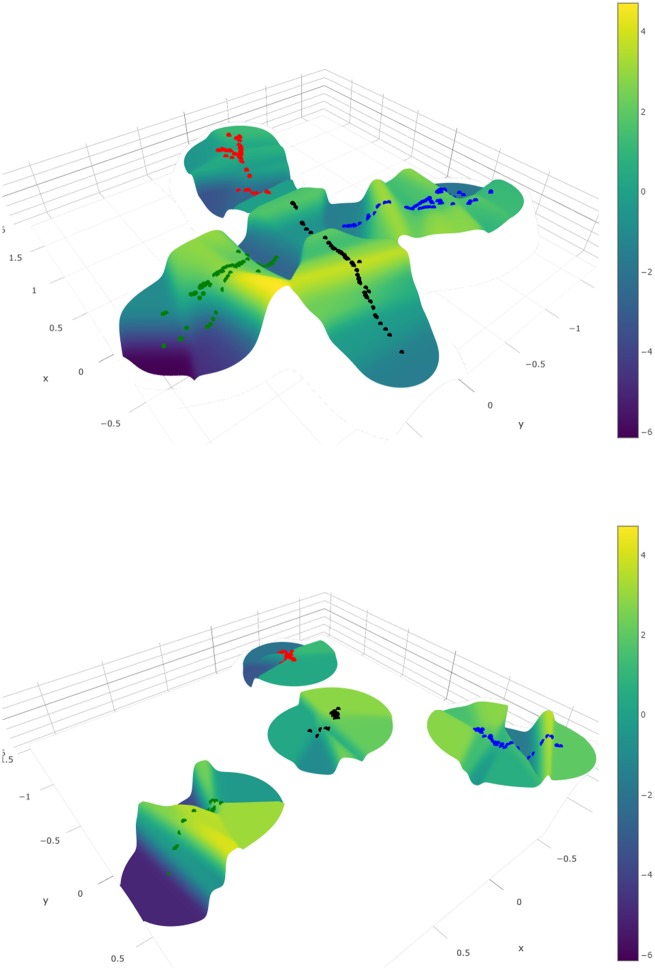
Rapidly evolving microbes are a challenge to model because of the volatile, complex, and dynamic nature of their populations. We developed the DISSEQT pipeline (DIStribution-based SEQuence space Time dynamics) for analyzing, visualizing, and predicting the evolution of heterogeneous biological populations in multidimensional genetic space, suited for population-based modeling of deep sequencing and high-throughput data. The pipeline is openly available on GitHub (https://github.com/rasmushenningsson/DISSEQT.jl, accessed 23 June 2019) and Synapse (https://www.synapse.org/#!Synapse: syn11425758, accessed 23 June 2019), covering the entire workflow from read alignment to visualization of results. Our pipeline is centered around robust dimension and model reduction algorithms for analysis of genotypic data with additional capabilities for including phenotypic features to explore dynamic genotype-phenotype maps. We illustrate its utility and capacity with examples from evolving RNA virus populations, which present one of the highest degrees of genetic heterogeneity within a given population found in nature. Using our pipeline, we empirically reconstruct the evolutionary trajectories of evolving populations in sequence space and genotype-phenotype fitness landscapes. We show that while sequence space is vastly multidimensional, the relevant genetic space of evolving microbial populations is of intrinsically low dimension. In addition, evolutionary trajectories of these populations can be faithfully monitored to identify the key minority genotypes contributing most to evolution. Finally, we show that empirical fitness landscapes, when reconstructed to include minority variants, can predict phenotype from genotype with high accuracy.
Keywords: NGS; applied mathematics; multidimensional scaling; quasispecies.
Figures

References
-
- Aronesty E. (2011) ‘ea-utils: Command-Line Tools for Processing Biological Sequencing Data’ <https://github.com/ExpressionAnalysis/ea-utils> accessed 23 June 2019.
LinkOut - more resources
Full Text Sources
