

Long-term outcome and unmet needs in infantile-onset Pompe disease
- PMID: 31392195
- PMCID: PMC6642934
- DOI: 10.21037/atm.2019.04.70
Long-term outcome and unmet needs in infantile-onset Pompe disease
Abstract
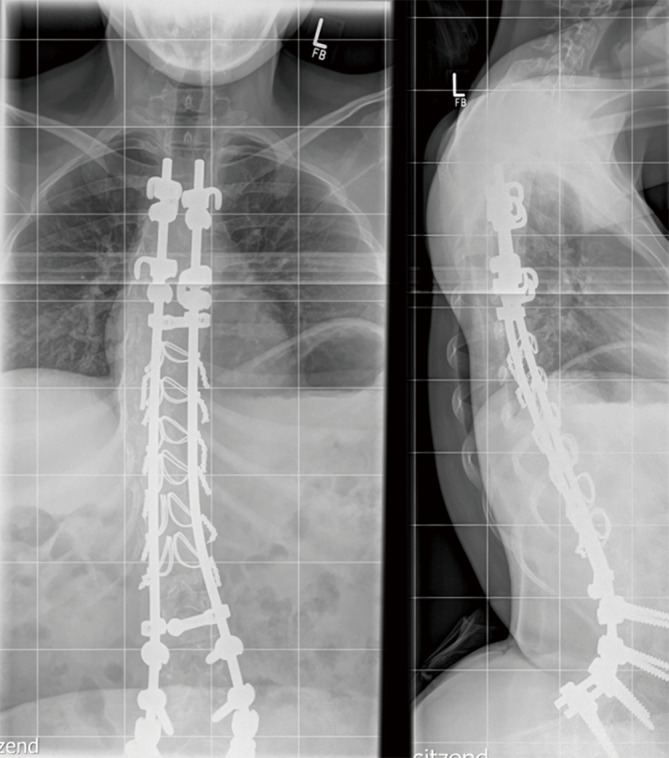
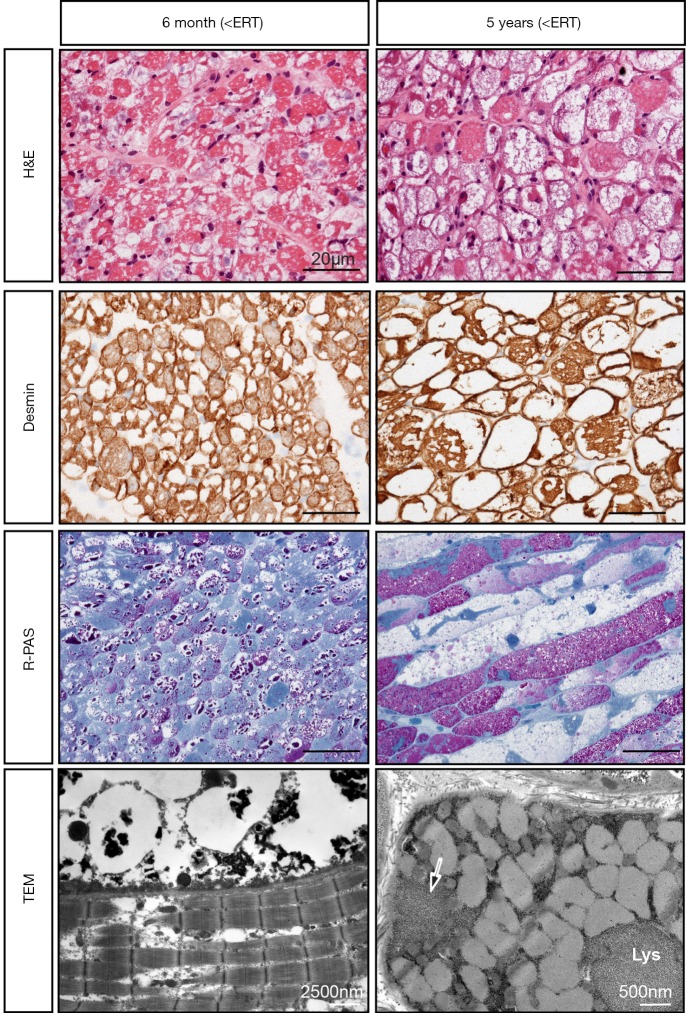
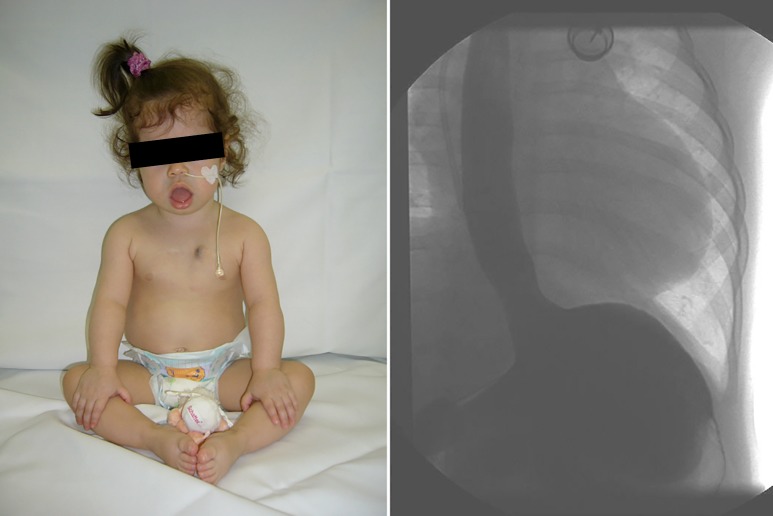
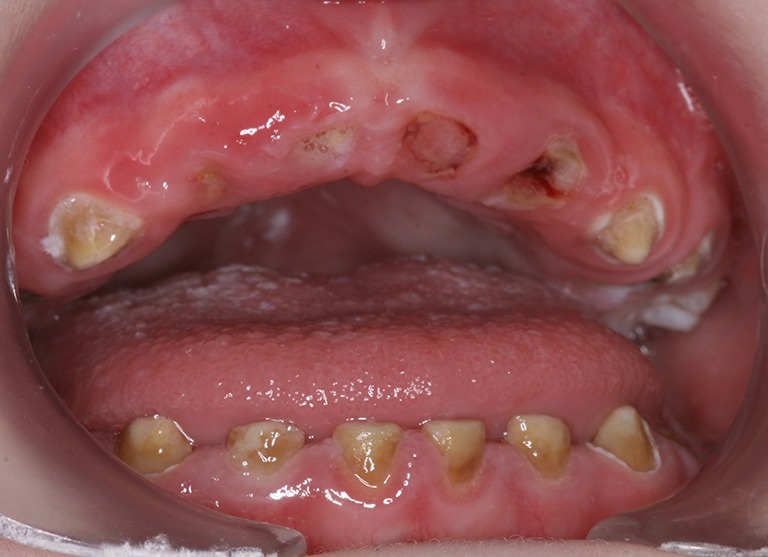
Infantile-onset Pompe disease (IOPD) is characterized by virtually complete absence of acid alpha-glucosidase (GAA)-activity, resulting in rapidly progressive hypertrophic cardiomyopathy (HCM), profound skeletal muscle weakness, and death usually within the first 12 months of life. Enzyme replacement therapy (ERT) with recombinant GAA in humans started in 1999, and pivotal studies demonstrated that the treatment ameliorated HCM, improved motor function in some patients, and prolonged overall and ventilator-free survival. These outcomes led to the approval of ERT in 2006. Implementation of ERT has uncovered multisystemic character of IOPD, not known in the pre-ERT era. Although ERT has substantially improved the prognosis of IOPD, mortality is still considerable, and decline of motor function with time is frequent in long-term survivors. This review details the new complex IOPD phenotype, outlines problems related to ERT, and highlights unmet needs.
Keywords: Infantile-onset Pompe disease (IOPD); enzyme replacement therapy (ERT); long-term outcome; musculoskeletal dysfunction; neurocognitive impairment.
Conflict of interest statement
Conflicts of Interest: The authors have no conflicts of interest to declare.
Figures
References
-
- Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver C, Beaudet A, Valle D, et al. editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill, 2001:3389-420.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous