

Disruption of the RICTOR/mTORC2 complex enhances the response of head and neck squamous cell carcinoma cells to PI3K inhibition
- PMID: 31393061
- PMCID: PMC6763779
- DOI: 10.1002/1878-0261.12558
Disruption of the RICTOR/mTORC2 complex enhances the response of head and neck squamous cell carcinoma cells to PI3K inhibition
Erratum in
-
Erratum to: Disruption of the RICTOR/mTORC2 complex enhances the response of head and neck squamous cell carcinoma cells to PI3K inhibition.Mol Oncol. 2020 Jan;14(1):230-231. doi: 10.1002/1878-0261.12604. Epub 2019 Dec 5. Mol Oncol. 2020. PMID: 31804024 Free PMC article. No abstract available.
Abstract
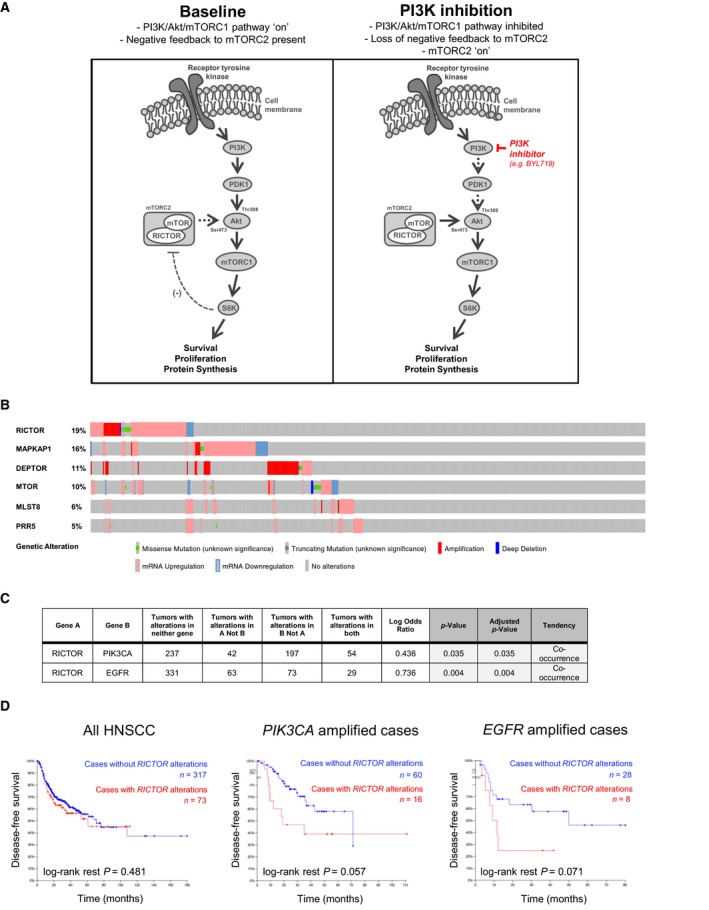
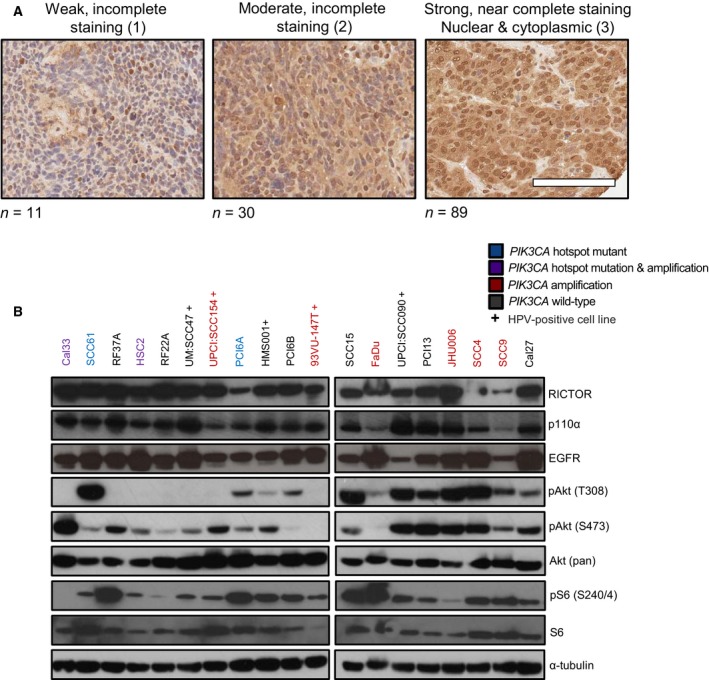
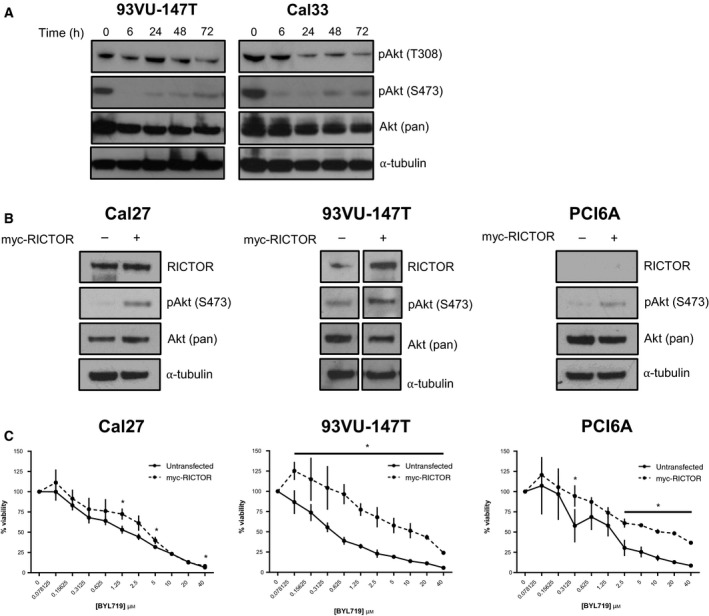
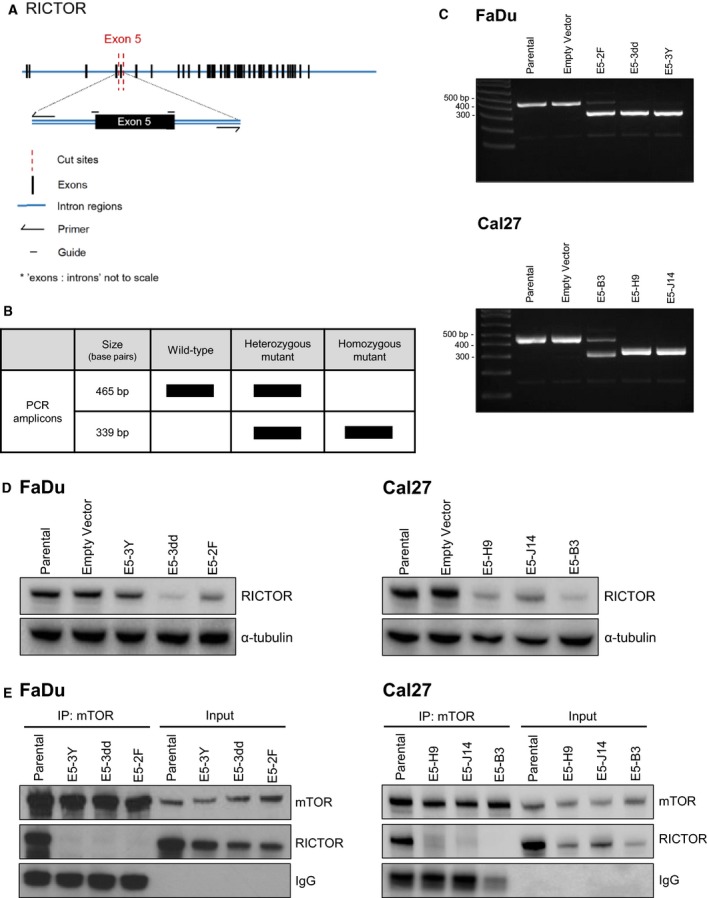
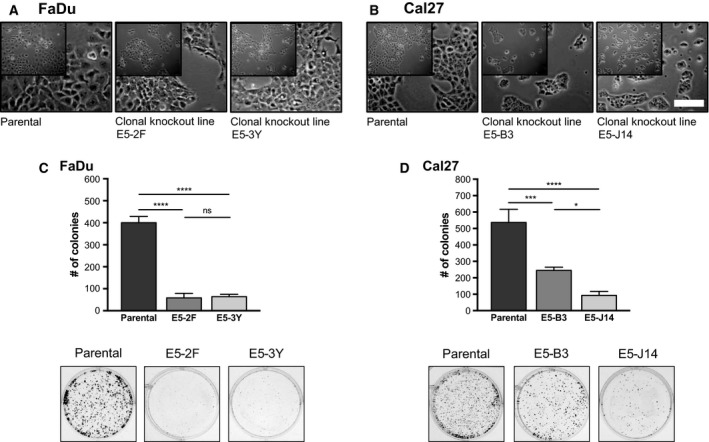
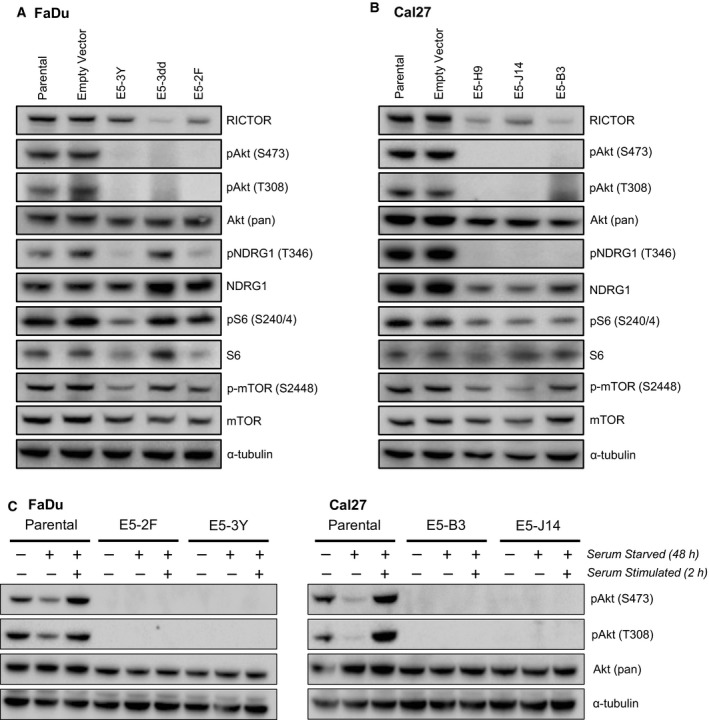
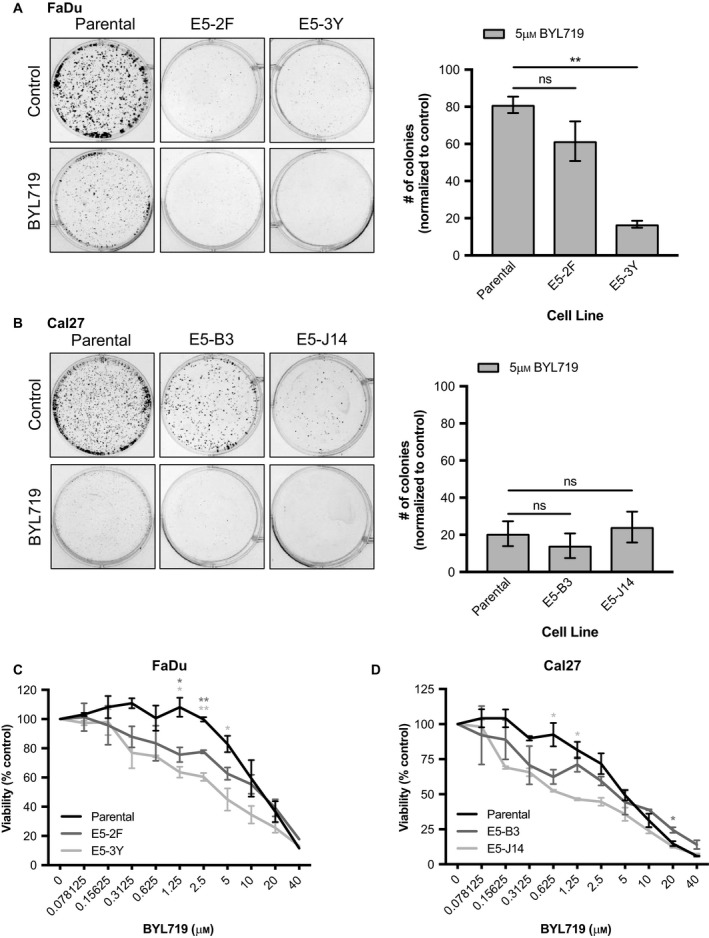
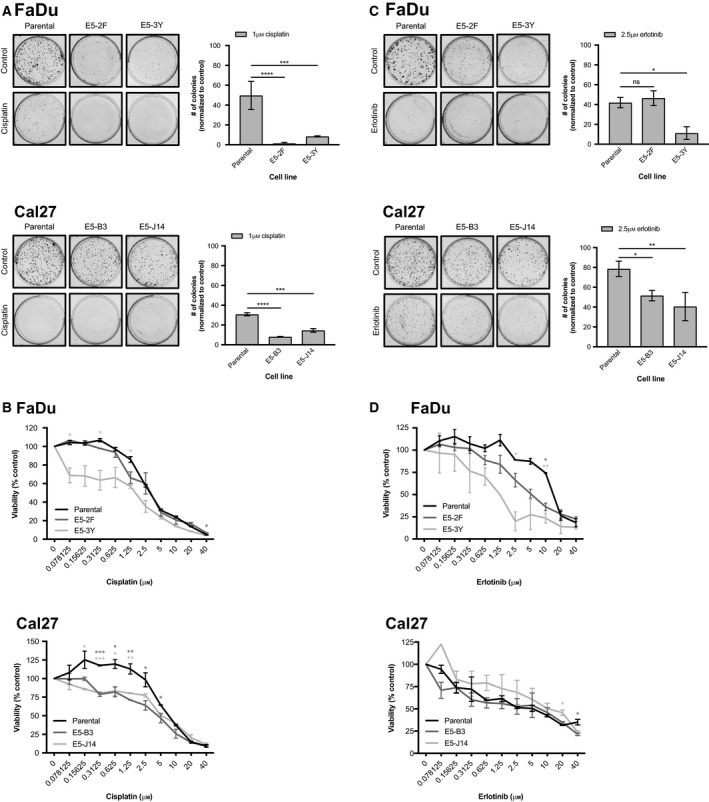
Phosphoinositide 3-kinase (PI3K) is aberrantly activated in head and neck squamous cell carcinomas (HNSCC) and plays a pivotal role in tumorigenesis by driving Akt signaling, leading to cell survival and proliferation. Phosphorylation of Akt Thr308 by PI3K-PDK1 and Akt Ser473 by mammalian target of rapamycin complex 2 (mTORC2) activates Akt. Targeted inhibition of PI3K is a major area of preclinical and clinical investigation as it reduces Akt Thr308 phosphorylation, suppressing downstream mTORC1 activity. However, inhibition of mTORC1 releases feedback inhibition of mTORC2, resulting in a resurgence of Akt activation mediated by mTORC2. While the role of PI3K-activated Akt signaling is well established in HNSCC, the significance of mTORC2-driven Akt signaling has not been thoroughly examined. Here we explore the expression and function of mTORC2 and its obligate subunit RICTOR in HNSCC primary tumors and cell lines. We find RICTOR to be overexpressed in a subset of HNSCC tumors, including those with PIK3CA or EGFR gene amplifications. Whereas overexpression of RICTOR reduced susceptibility of HNSCC tumor cells to PI3K inhibition, genetic ablation of RICTOR using CRISPR/Cas9 sensitized cells to PI3K inhibition, as well as to EGFR inhibition and cisplatin treatment. Further, mTORC2 disruption led to reduced viability and colony forming abilities of HNSCC cells relative to their parental lines and induced loss of both activating Akt phosphorylation modifications (Thr308 and Ser473). Taken together, our findings establish RICTOR/mTORC2 as a critical oncogenic complex in HNSCC and rationalize the development of an mTORC2-specific inhibitor for use in HNSCC, either combined with agents already under investigation, or as an independent therapy.
Keywords: PI3-kinase; RICTOR; head and neck cancer; mTORC2; targeted therapy.
© 2019 The Authors. Published by FEBS Press and John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
