

ALG9 Mutation Carriers Develop Kidney and Liver Cysts
- PMID: 31395617
- PMCID: PMC6830805
- DOI: 10.1681/ASN.2019030298
ALG9 Mutation Carriers Develop Kidney and Liver Cysts
Abstract
Background: Mutations in PKD1 or PKD2 cause typical autosomal dominant polycystic kidney disease (ADPKD), the most common monogenic kidney disease. Dominantly inherited polycystic kidney and liver diseases on the ADPKD spectrum are also caused by mutations in at least six other genes required for protein biogenesis in the endoplasmic reticulum, the loss of which results in defective production of the PKD1 gene product, the membrane protein polycystin-1 (PC1).
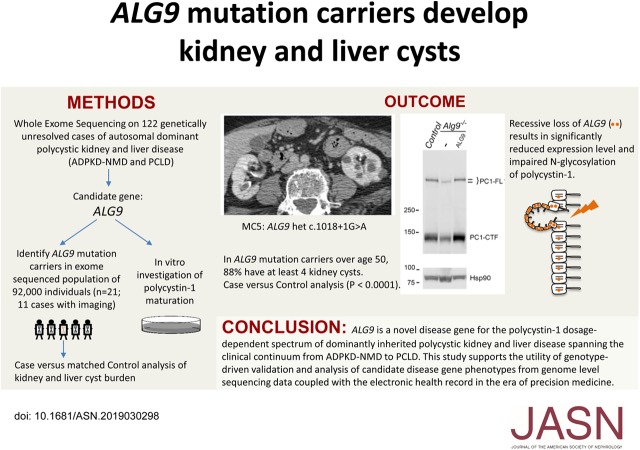
Methods: We used whole-exome sequencing in a cohort of 122 patients with genetically unresolved clinical diagnosis of ADPKD or polycystic liver disease to identify a candidate gene, ALG9, and in vitro cell-based assays of PC1 protein maturation to functionally validate it. For further validation, we identified carriers of ALG9 loss-of-function mutations and noncarrier matched controls in a large exome-sequenced population-based cohort and evaluated the occurrence of polycystic phenotypes in both groups.
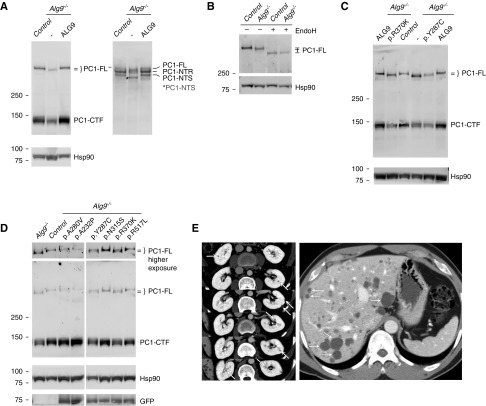
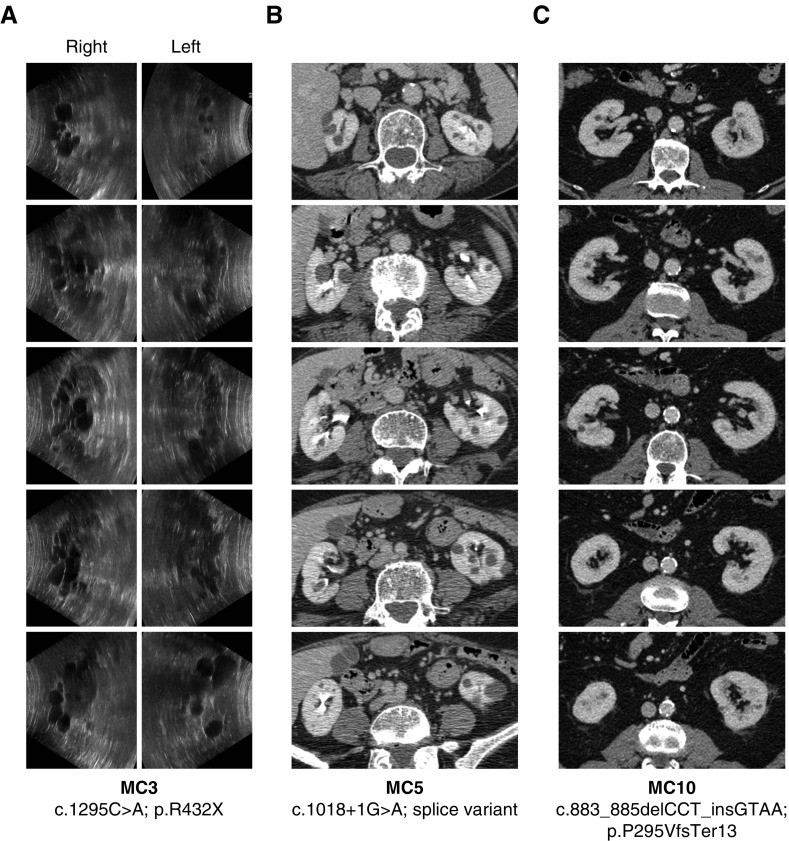
Results: Two patients in the clinically defined cohort had rare loss-of-function variants in ALG9, which encodes a protein required for addition of specific mannose molecules to the assembling N-glycan precursors in the endoplasmic reticulum lumen. In vitro assays showed that inactivation of Alg9 results in impaired maturation and defective glycosylation of PC1. Seven of the eight (88%) cases selected from the population-based cohort based on ALG9 mutation carrier state who had abdominal imaging after age 50; seven (88%) had at least four kidney cysts, compared with none in matched controls without ALG9 mutations.
Conclusions: ALG9 is a novel disease gene in the genetically heterogeneous ADPKD spectrum. This study supports the utility of phenotype characterization in genetically-defined cohorts to validate novel disease genes, and provide much-needed genotype-phenotype correlations.
Keywords: ADPKD; chronic renal disease; cystic kidney; human genetics; kidney stones; polycystic kidney disease.
Copyright © 2019 by the American Society of Nephrology.
Figures
Comment in
-
New Ways of Finding New Genes for Old Diseases.J Am Soc Nephrol. 2019 Nov;30(11):2037-2039. doi: 10.1681/ASN.2019090940. Epub 2019 Oct 10. J Am Soc Nephrol. 2019. PMID: 31601610 Free PMC article. No abstract available.
References
-
- Torres VE, Harris PC, Pirson Y: Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007 - PubMed
-
- Drenth JP, te Morsche RH, Smink R, Bonifacino JS, Jansen JB: Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat Genet 33: 345–347, 2003 - PubMed
-
- Davila S, Furu L, Gharavi AG, Tian X, Onoe T, Qian Q, et al.: Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat Genet 36: 575–577, 2004 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
