

Alchemical Grid Dock (AlGDock): Binding Free Energy Calculations between Flexible Ligands and Rigid Receptors
- PMID: 31397498
- PMCID: PMC7263302
- DOI: 10.1002/jcc.26036
Alchemical Grid Dock (AlGDock): Binding Free Energy Calculations between Flexible Ligands and Rigid Receptors
Abstract
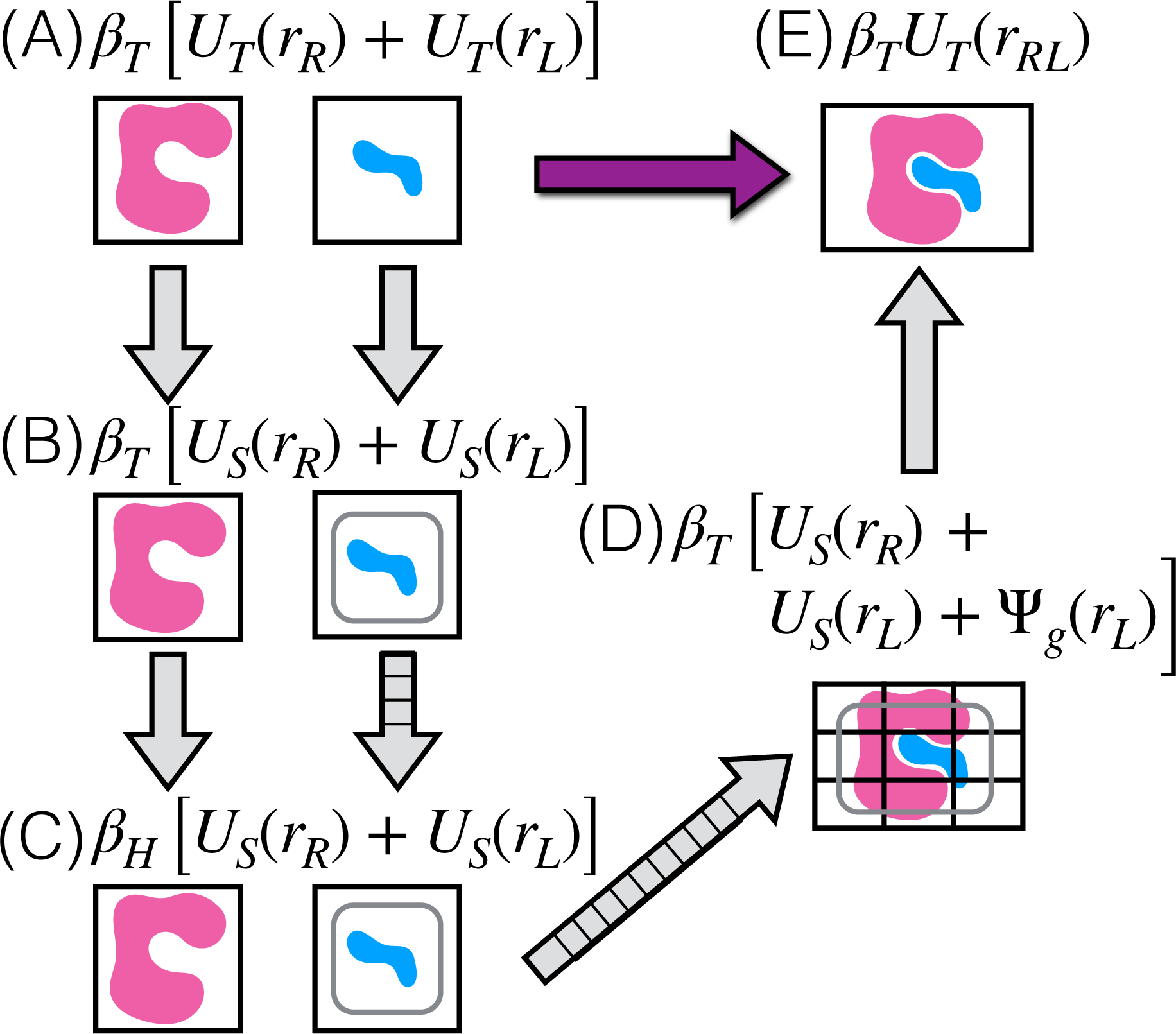
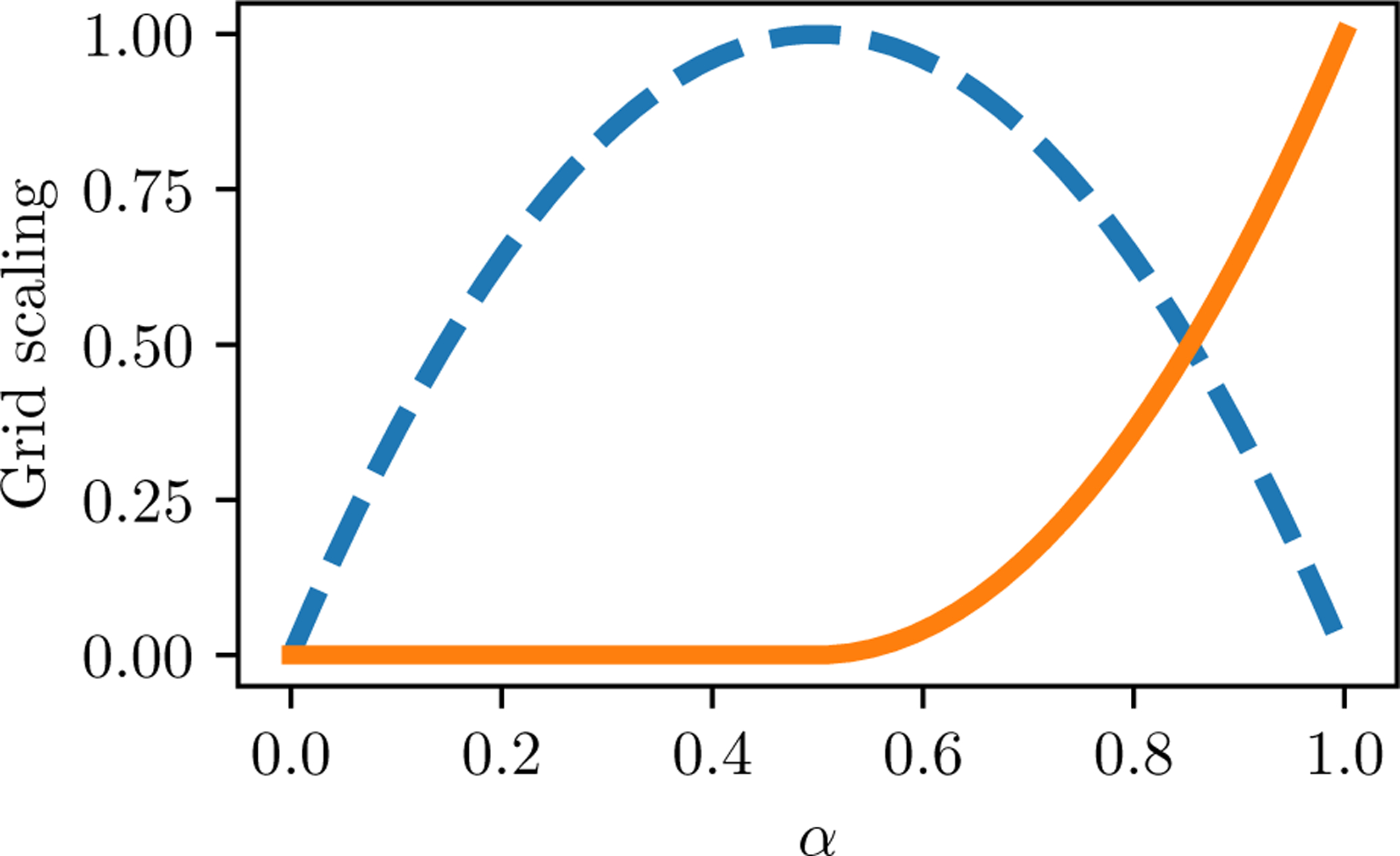
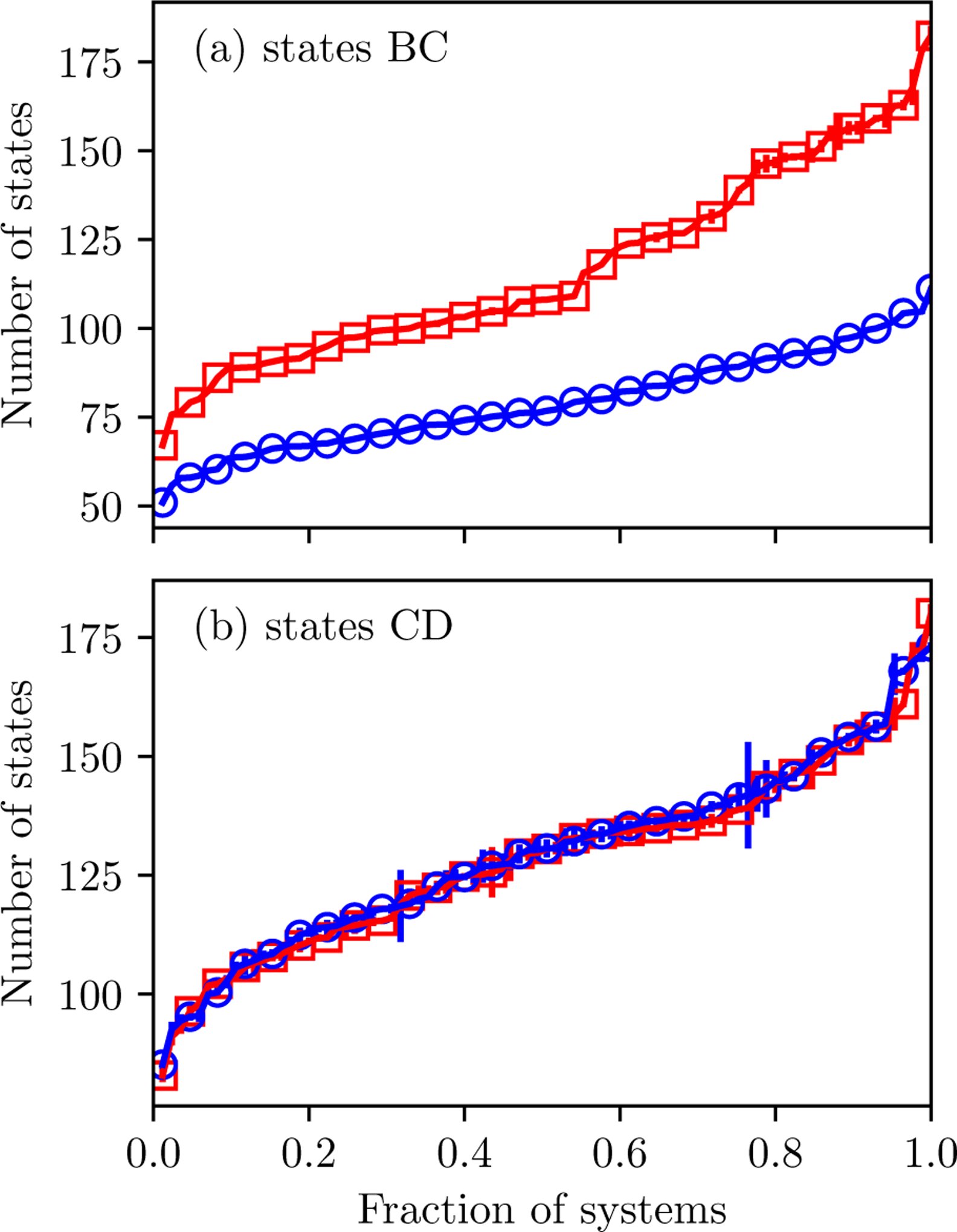
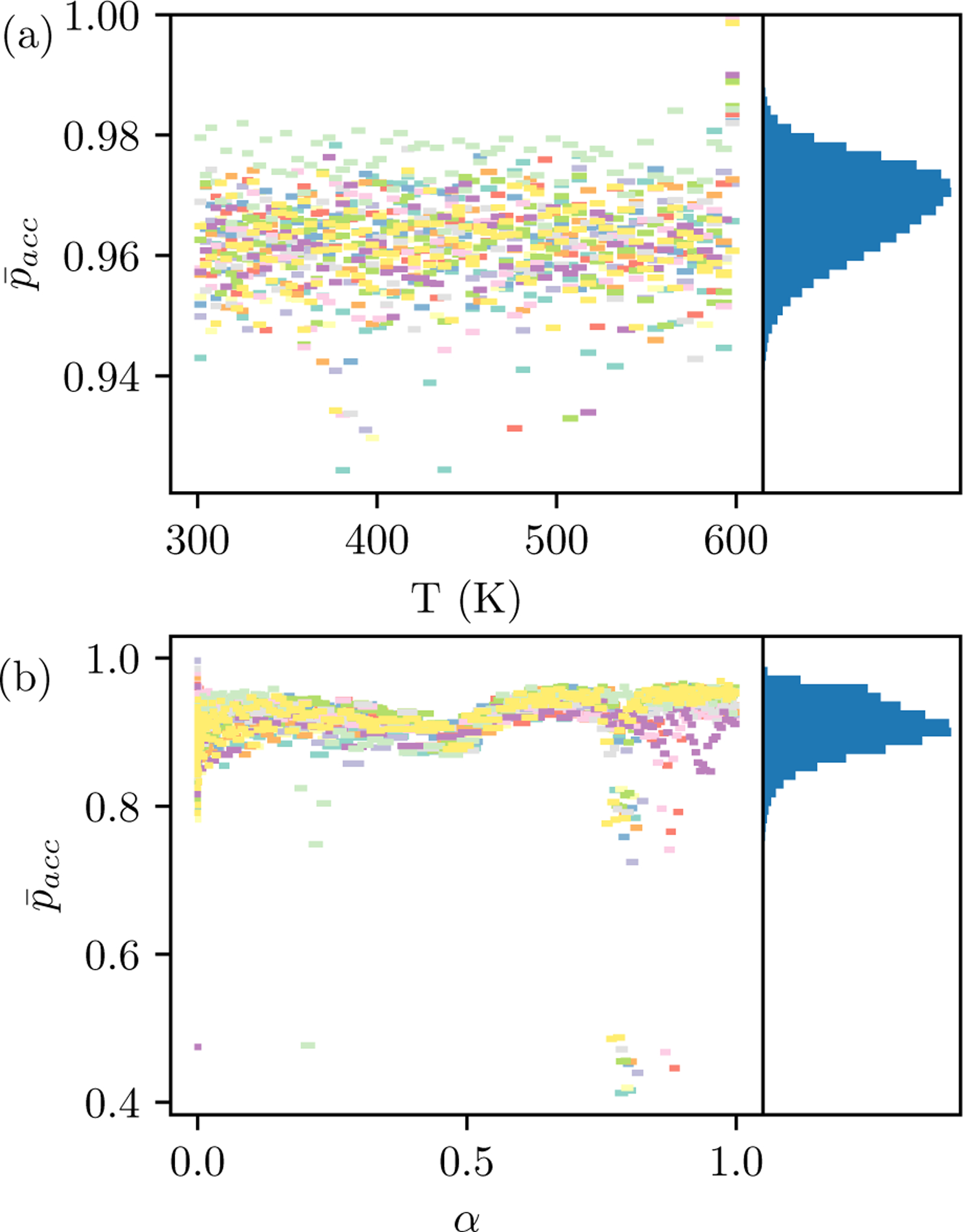
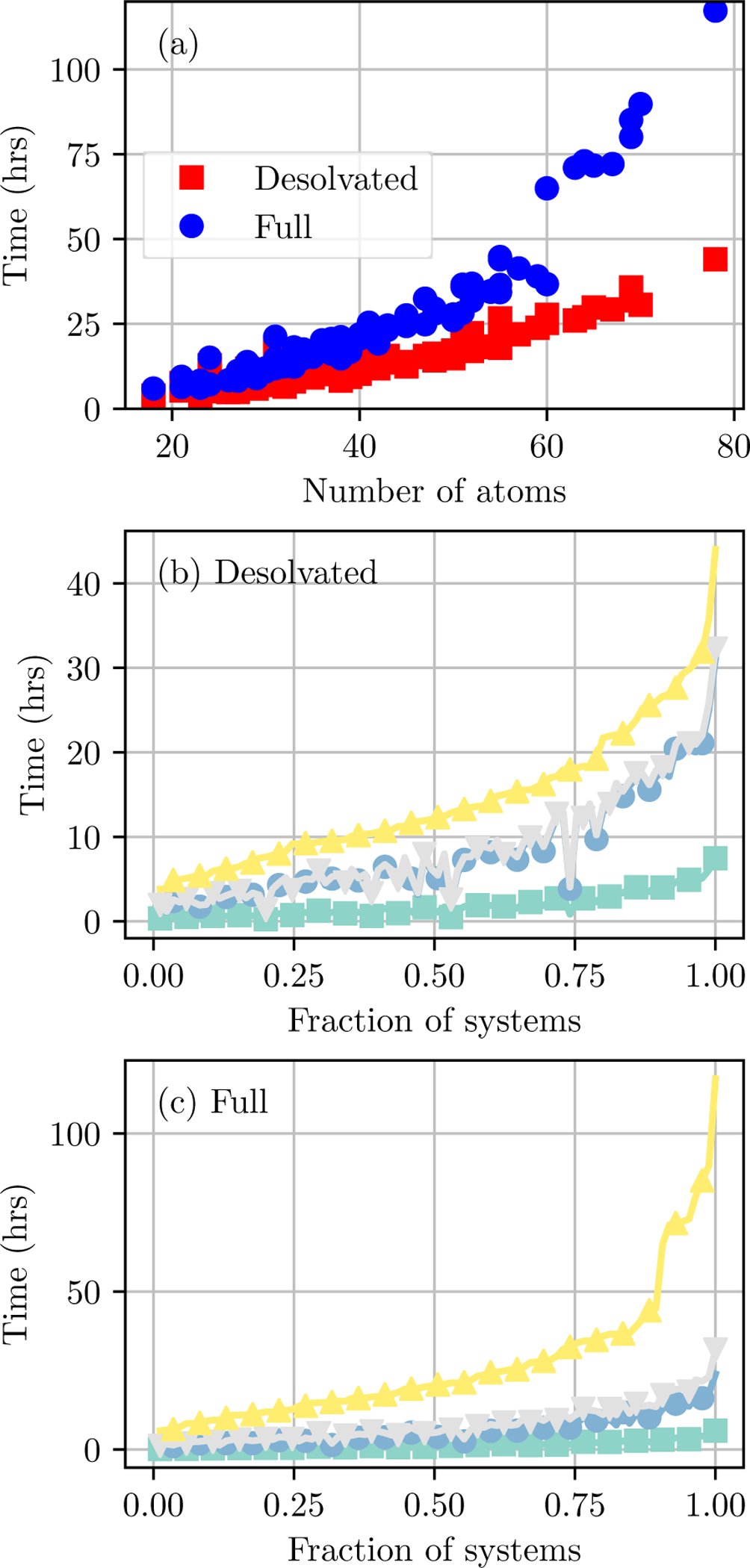
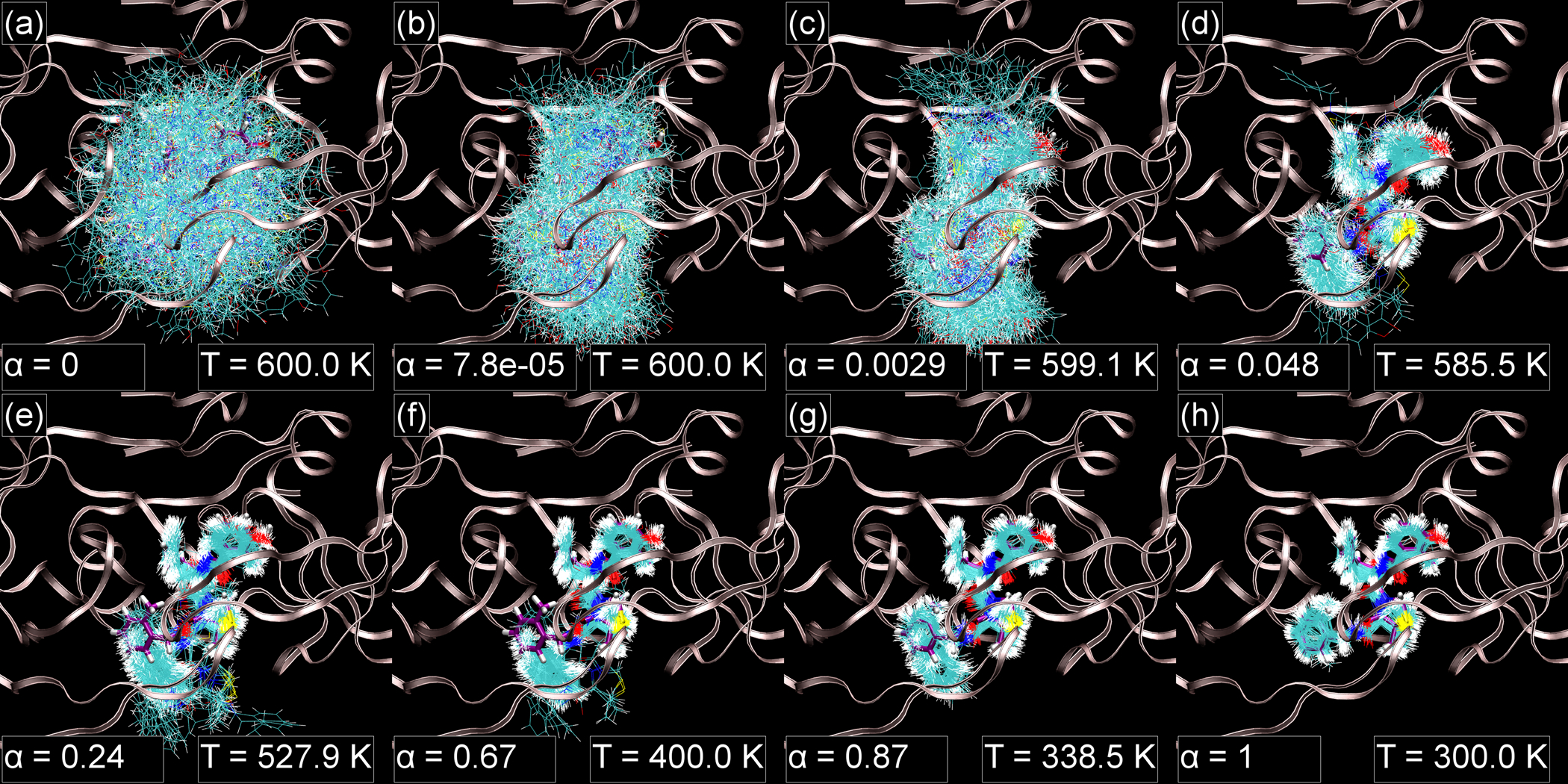
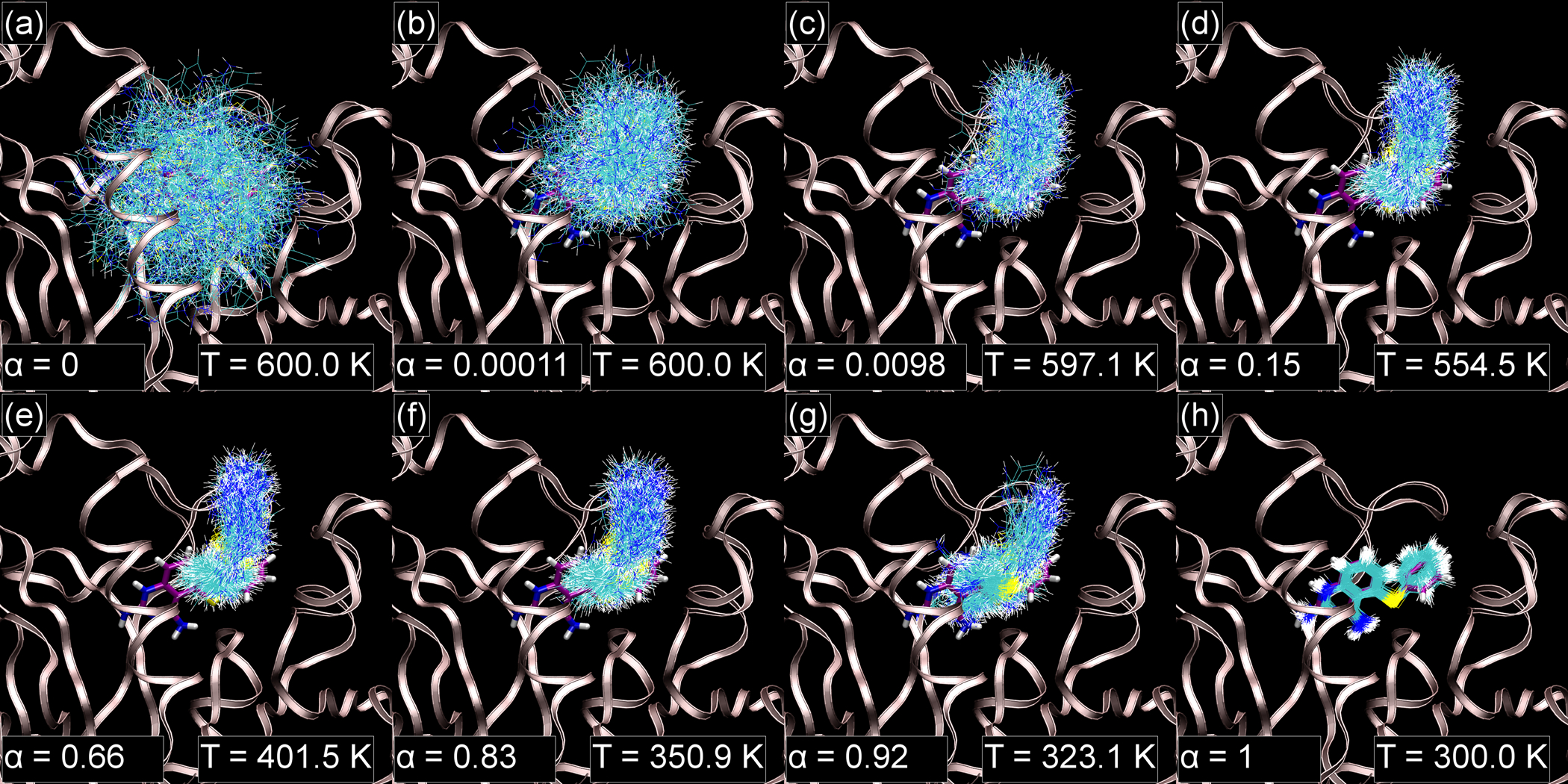
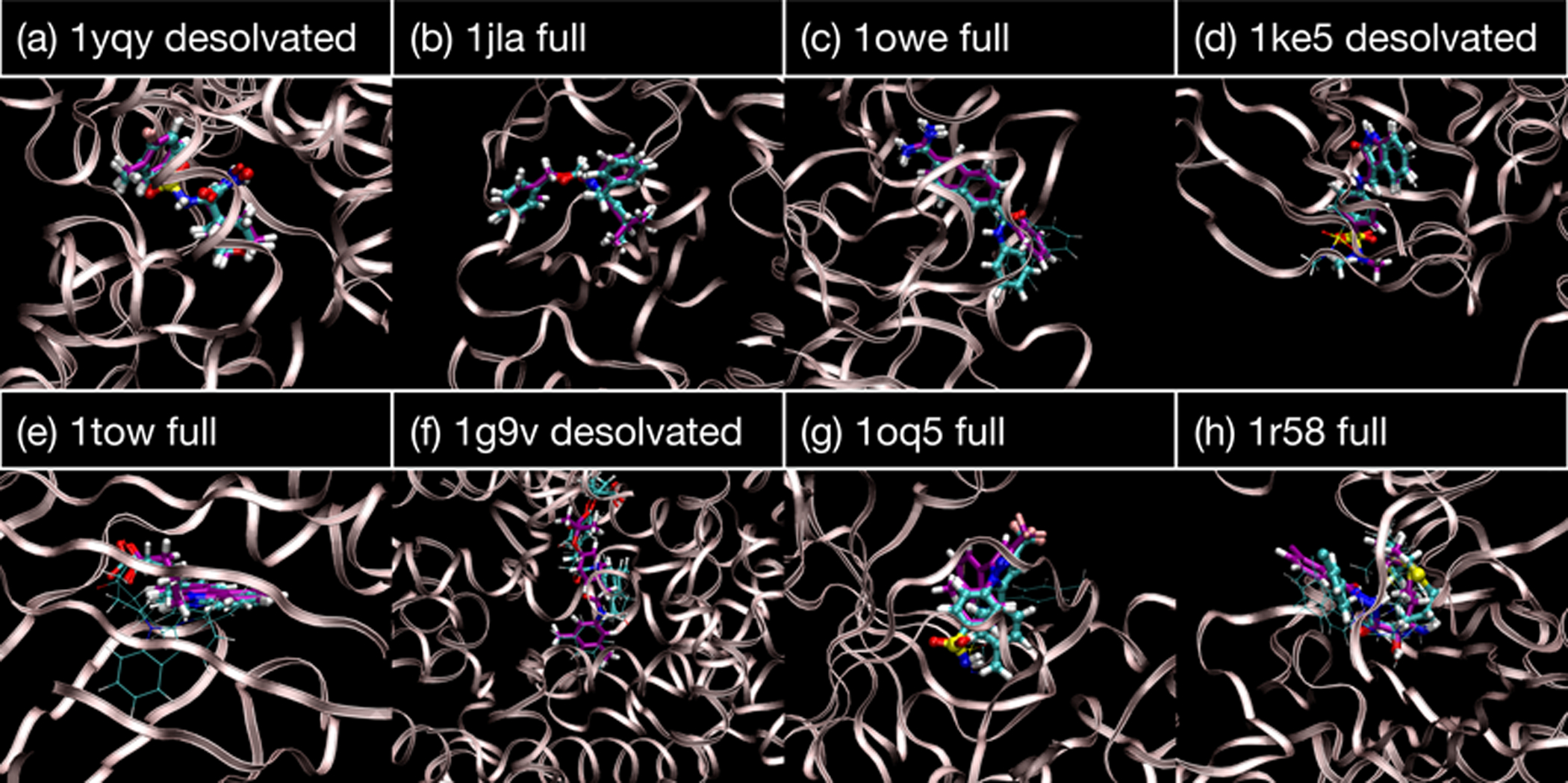
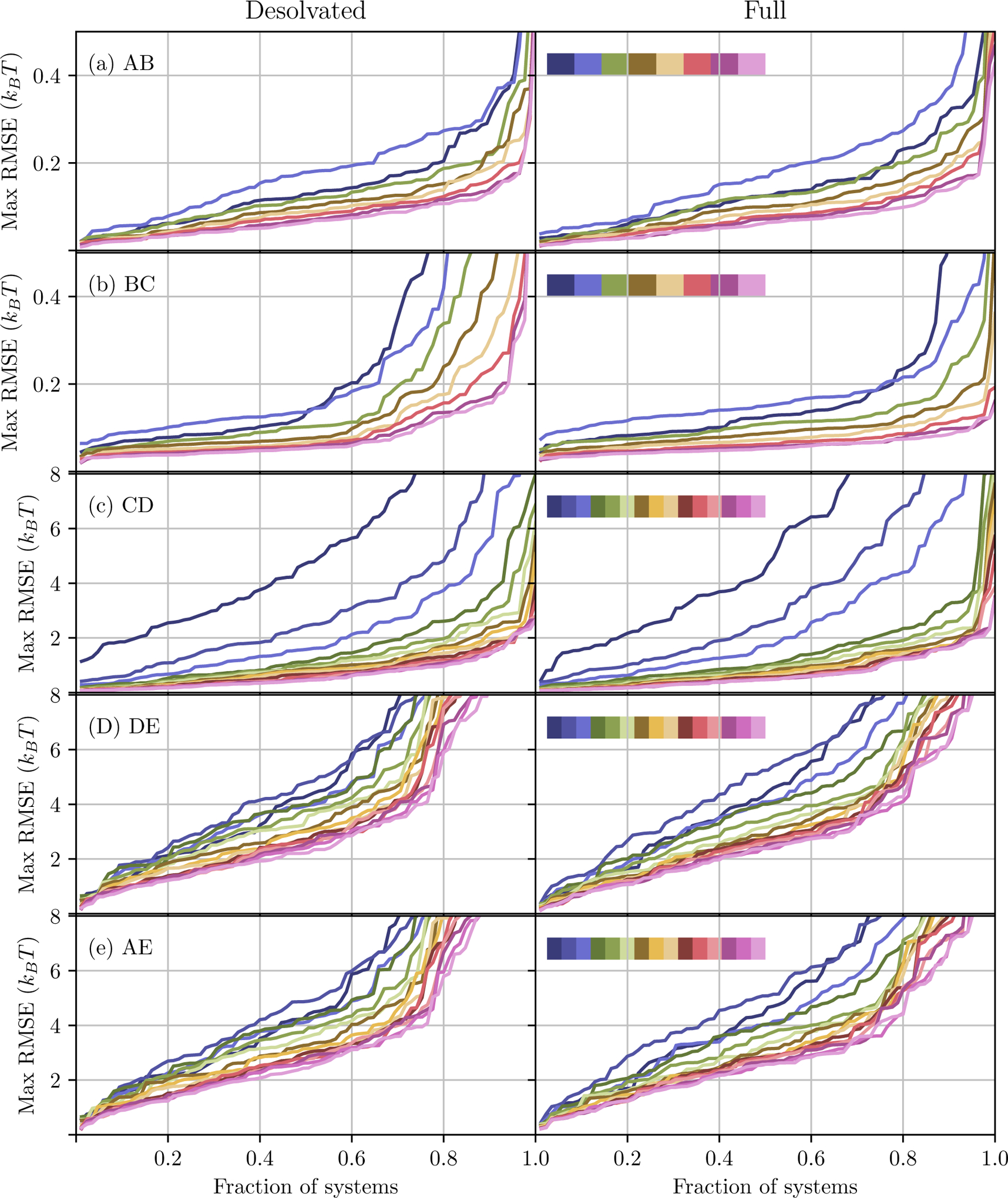
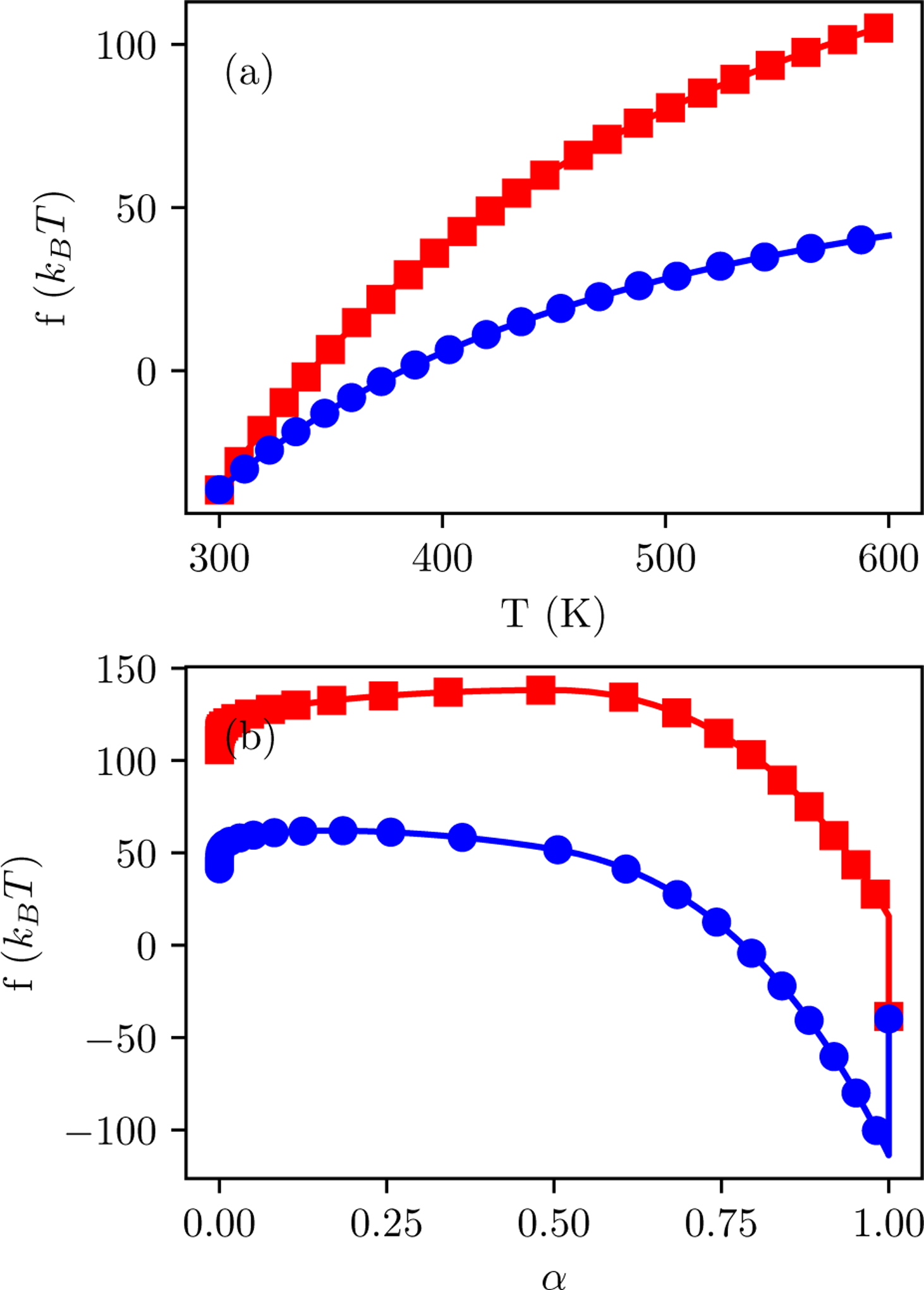
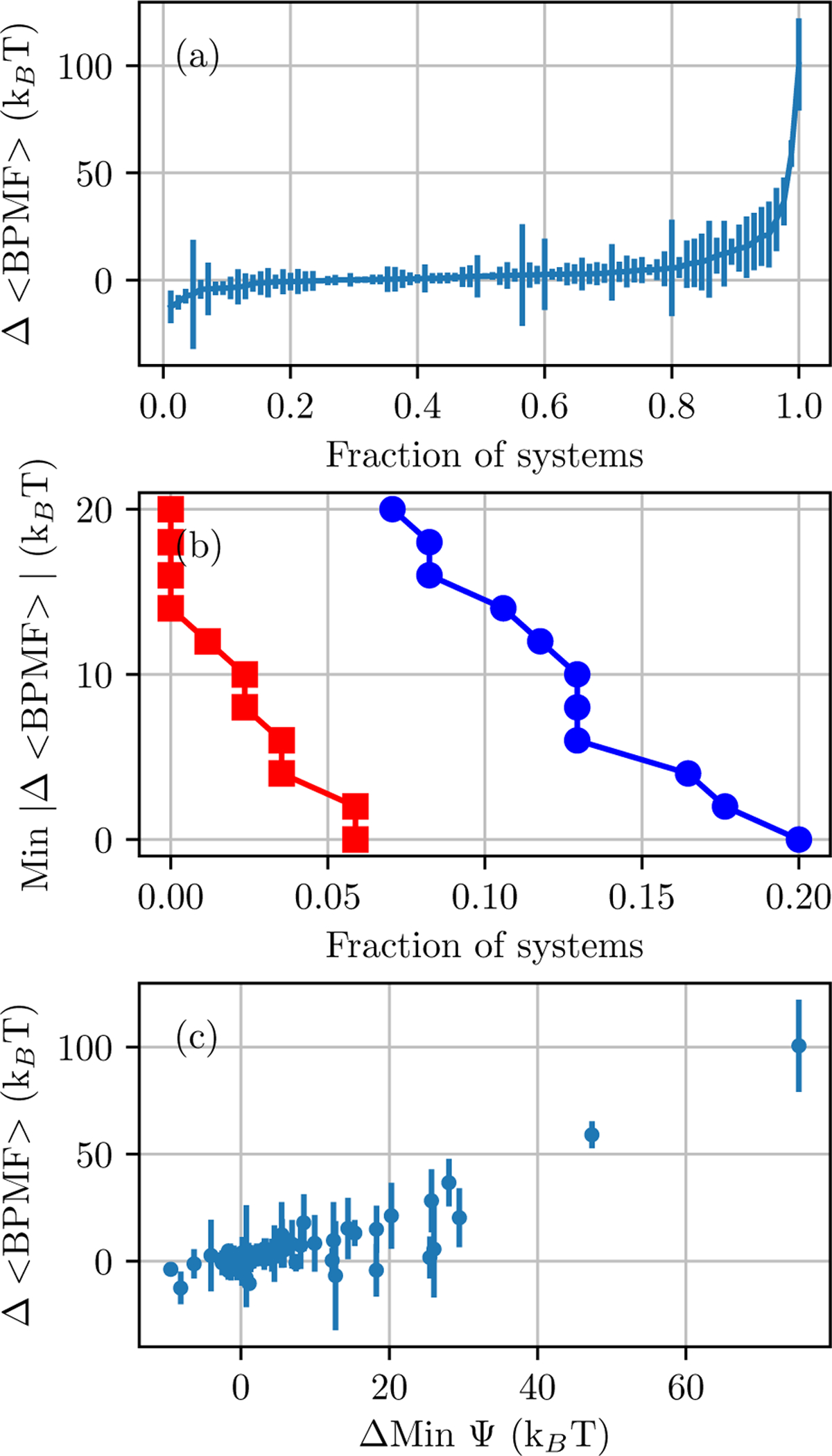
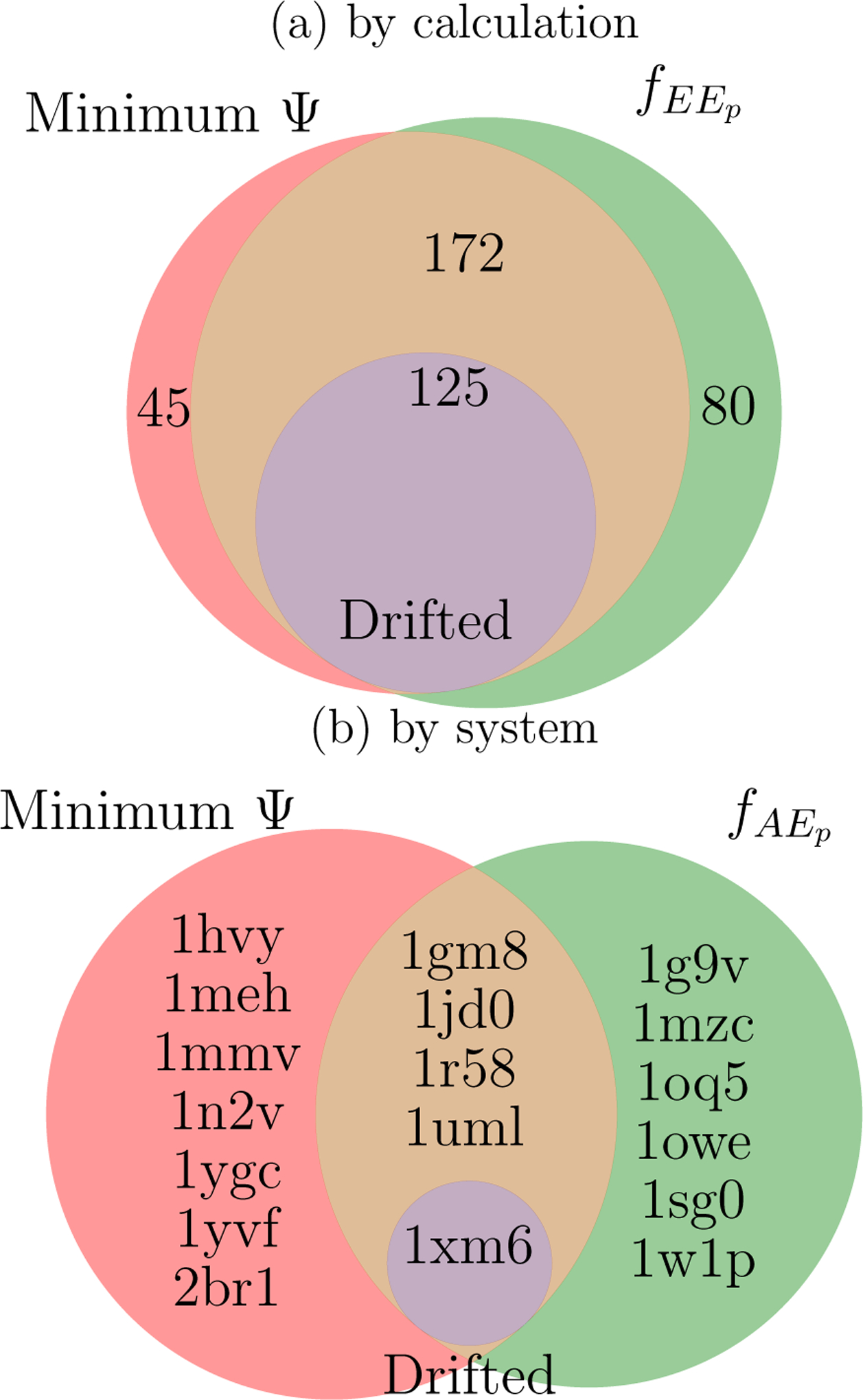
Alchemical Grid Dock (AlGDock) is open-source software designed to compute the binding potential of mean force-the binding free energy between a flexible ligand and a rigid receptor-for a small organic ligand and a biological macromolecule. Multiple BPMFs can be used to rigorously compute binding affinities between flexible partners. AlGDock uses replica exchange between thermodynamic states at different temperatures and receptor-ligand interaction strengths. Receptor-ligand interaction energies are represented by interpolating precomputed grids. Thermodynamic states are adaptively initialized and adjusted on-the-fly to maintain adequate replica exchange rates. In demonstrative calculations, when the bound ligand is treated as fully solvated, AlGDock estimates BPMFs with a precision within 4 kT in 65% and within 8 kT for 91% of systems. It correctly identifies the native binding pose in 83% of simulations. Performance is sometimes limited by subtle differences in the important configuration space of sampled and targeted thermodynamic states. © 2019 Wiley Periodicals, Inc.
Keywords: implicit ligand theory; noncovalent binding free energy; protein-ligand; replica exchange; thermodynamic length.
© 2019 Wiley Periodicals, Inc.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
