

Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas
- PMID: 31398252
- PMCID: PMC6954438
- DOI: 10.1093/neuonc/noz140
Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas
Abstract
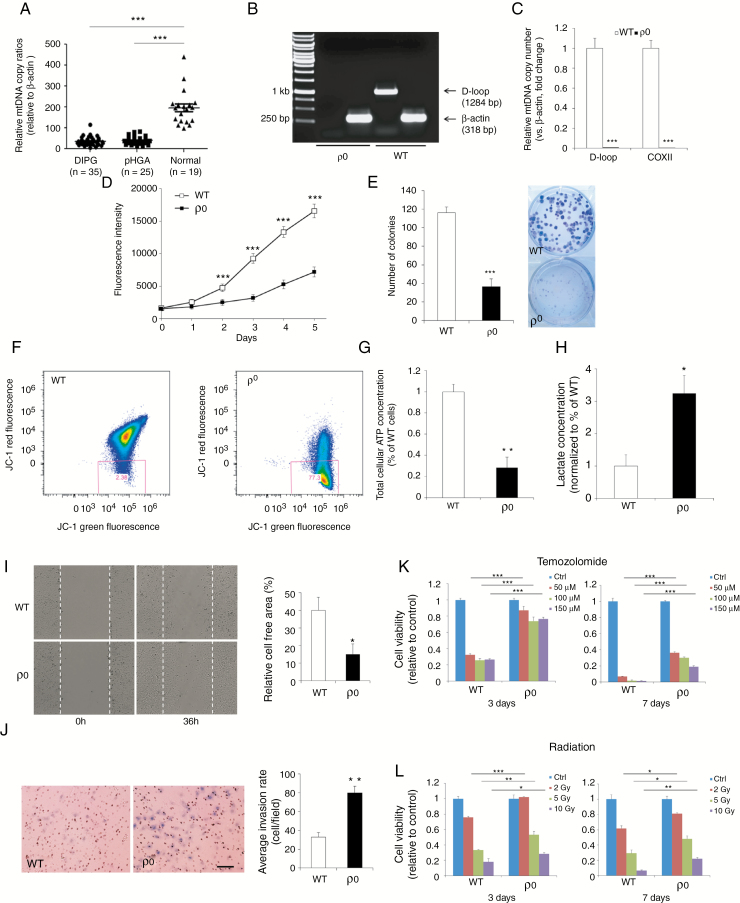
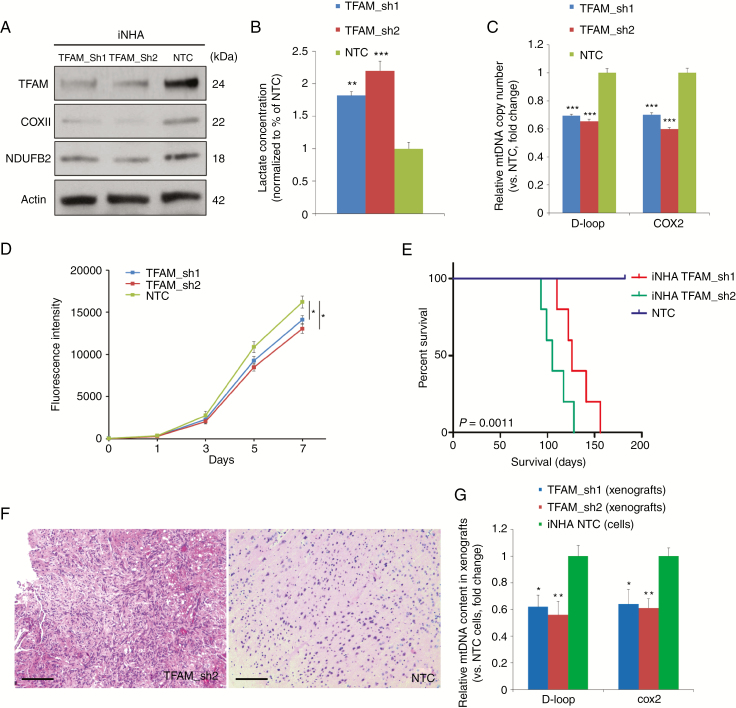
Background: Despite increased understanding of the genetic events underlying pediatric high-grade gliomas (pHGGs), therapeutic progress is static, with poor understanding of nongenomic drivers. We therefore investigated the role of alterations in mitochondrial function and developed an effective combination therapy against pHGGs.
Methods: Mitochondrial DNA (mtDNA) copy number was measured in a cohort of 60 pHGGs. The implication of mtDNA alteration in pHGG tumorigenesis was studied and followed by an efficacy investigation using patient-derived cultures and orthotopic xenografts.
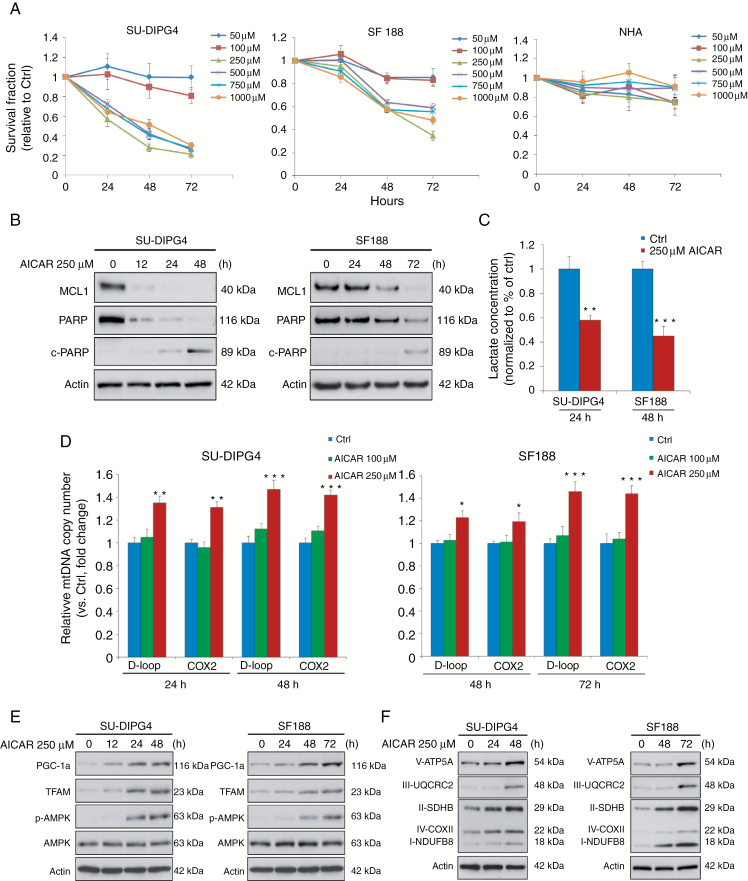
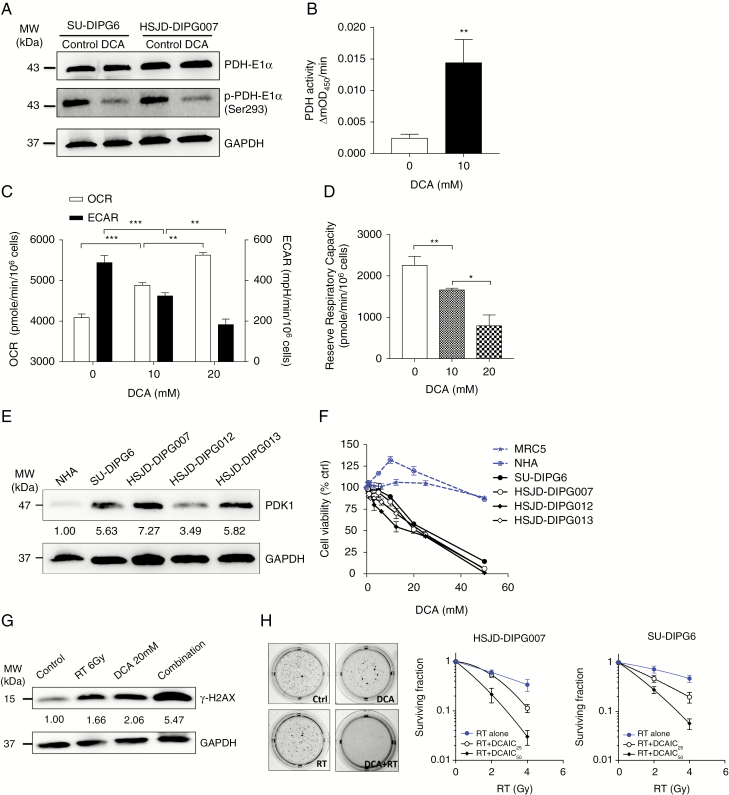
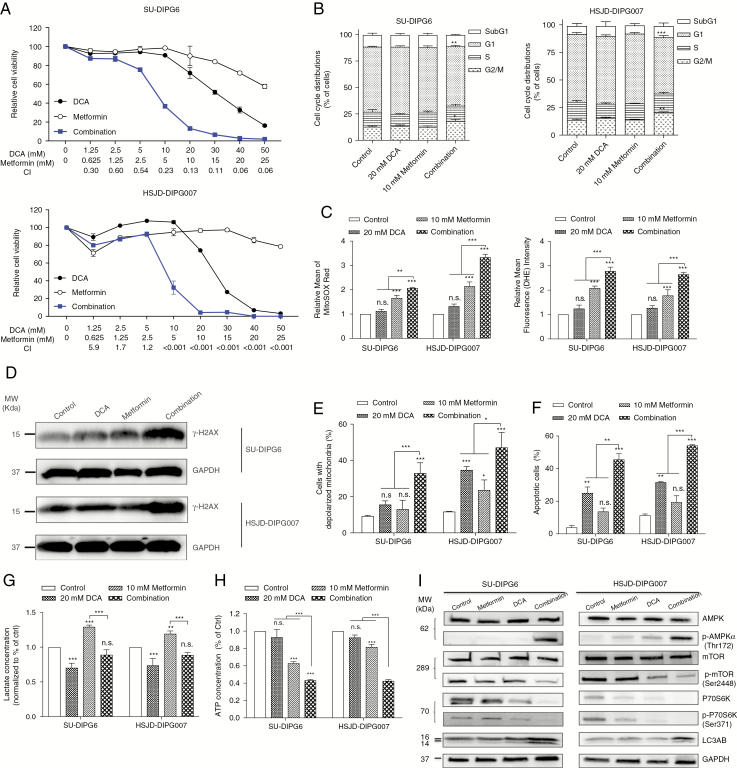
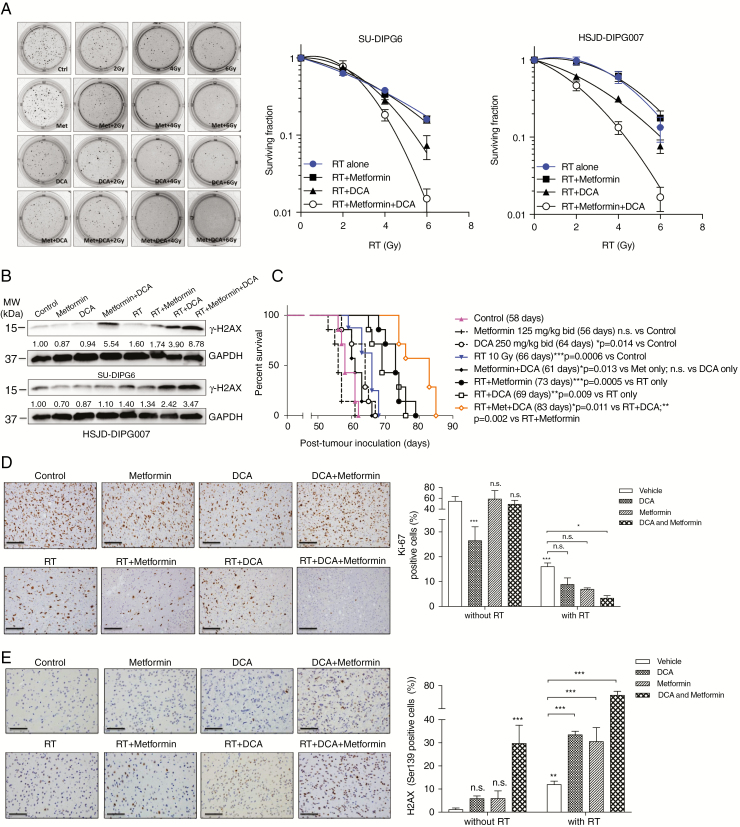
Results: Average mtDNA content was significantly lower in tumors versus normal brains. Decreasing mtDNA copy number in normal human astrocytes led to a markedly increased tumorigenicity in vivo. Depletion of mtDNA in pHGG cells promoted cell migration and invasion and therapeutic resistance. Shifting glucose metabolism from glycolysis to mitochondrial oxidation with the adenosine monophosphate-activated protein kinase activator AICAR (5-aminoimidazole-4-carboxamide ribonucleotide) or the pyruvate dehydrogenase kinase inhibitor dichloroacetate (DCA) significantly inhibited pHGG viability. Using DCA to shift glucose metabolism to mitochondrial oxidation and then metformin to simultaneously target mitochondrial function disrupted energy homeostasis of tumor cells, increasing DNA damage and apoptosis. The triple combination with radiation therapy, DCA and metformin led to a more potent therapeutic effect in vitro and in vivo.
Conclusions: Our results suggest metabolic alterations as an onco-requisite factor of pHGG tumorigenesis. Targeting reduced mtDNA quantity represents a promising therapeutic strategy for pHGG.
Keywords: DIPG; gliomas; mitochondria; radiotherapy.
© The Author(s) 2019. Published by Oxford University Press on behalf of the Society for Neuro-Oncology. For commercial re-use, please contact journals.permissions@oup.com.
Figures
References
-
- Yu M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011;89(3-4):65–71. - PubMed
-
- Sattler UG, Meyer SS, Quennet V, et al. . Glycolytic metabolism and tumour response to fractionated irradiation. Radiother Oncol. 2010;94(1):102–109. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
