

Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks
- PMID: 31398340
- PMCID: PMC7102900
- DOI: 10.1016/j.cell.2019.07.015
Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks
Abstract
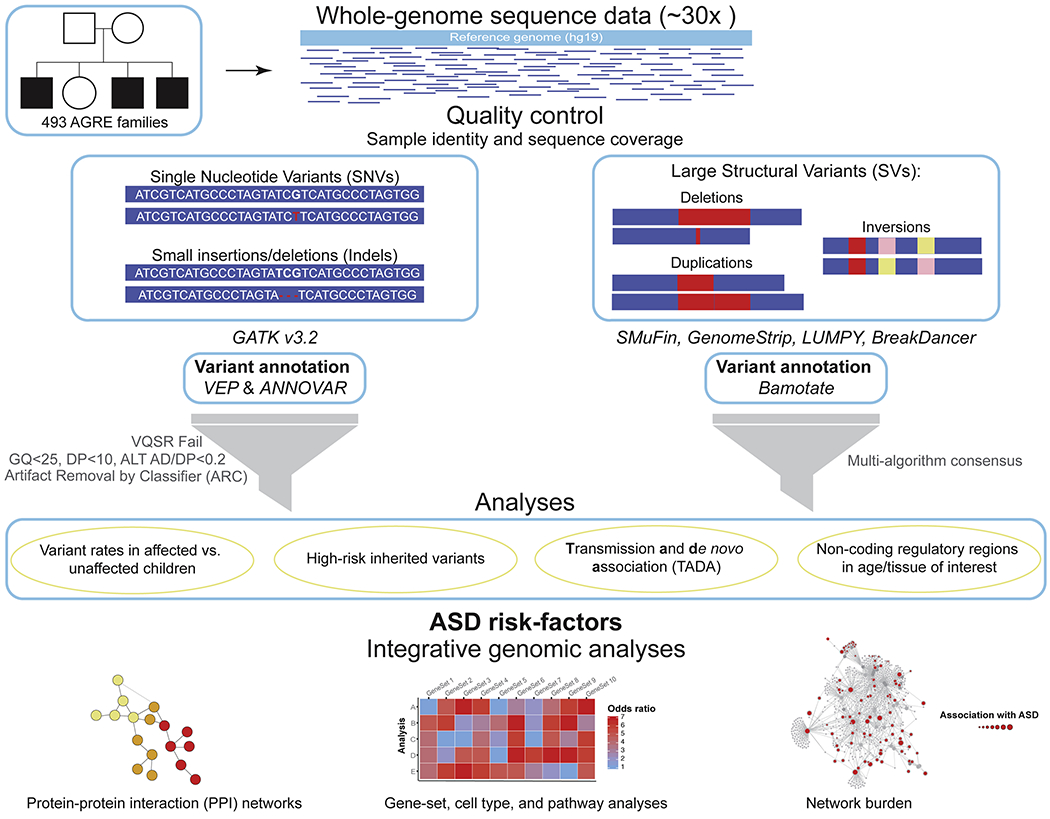
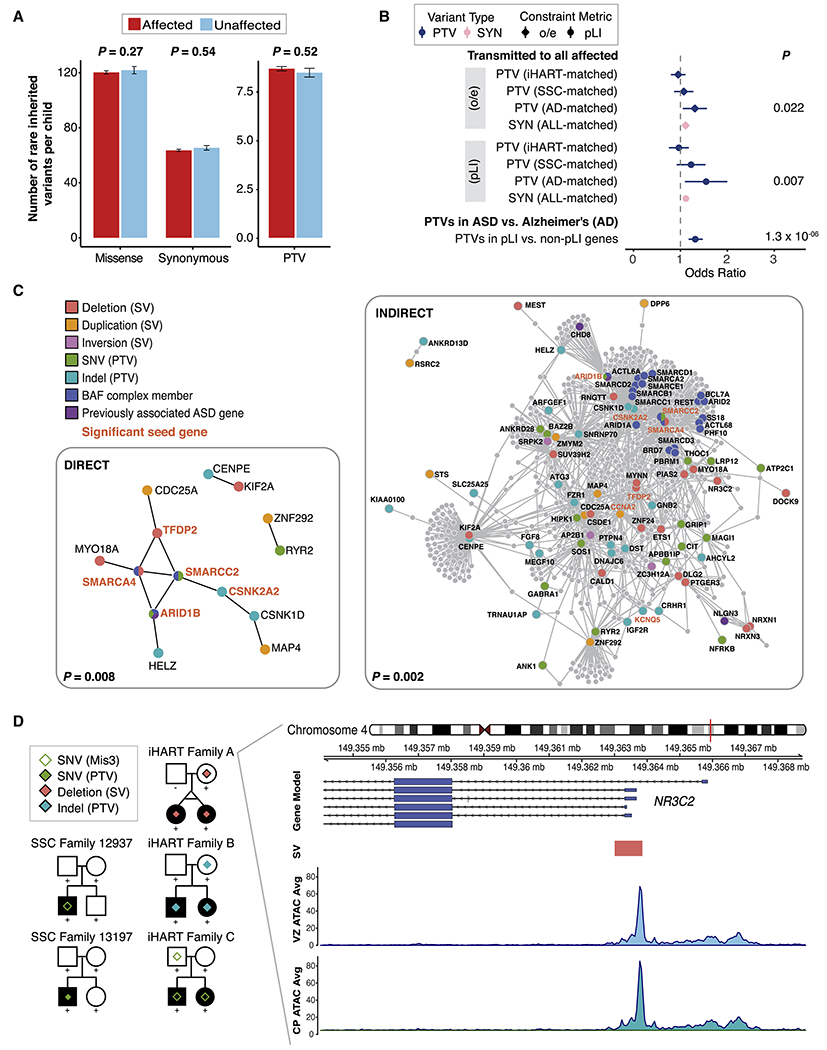
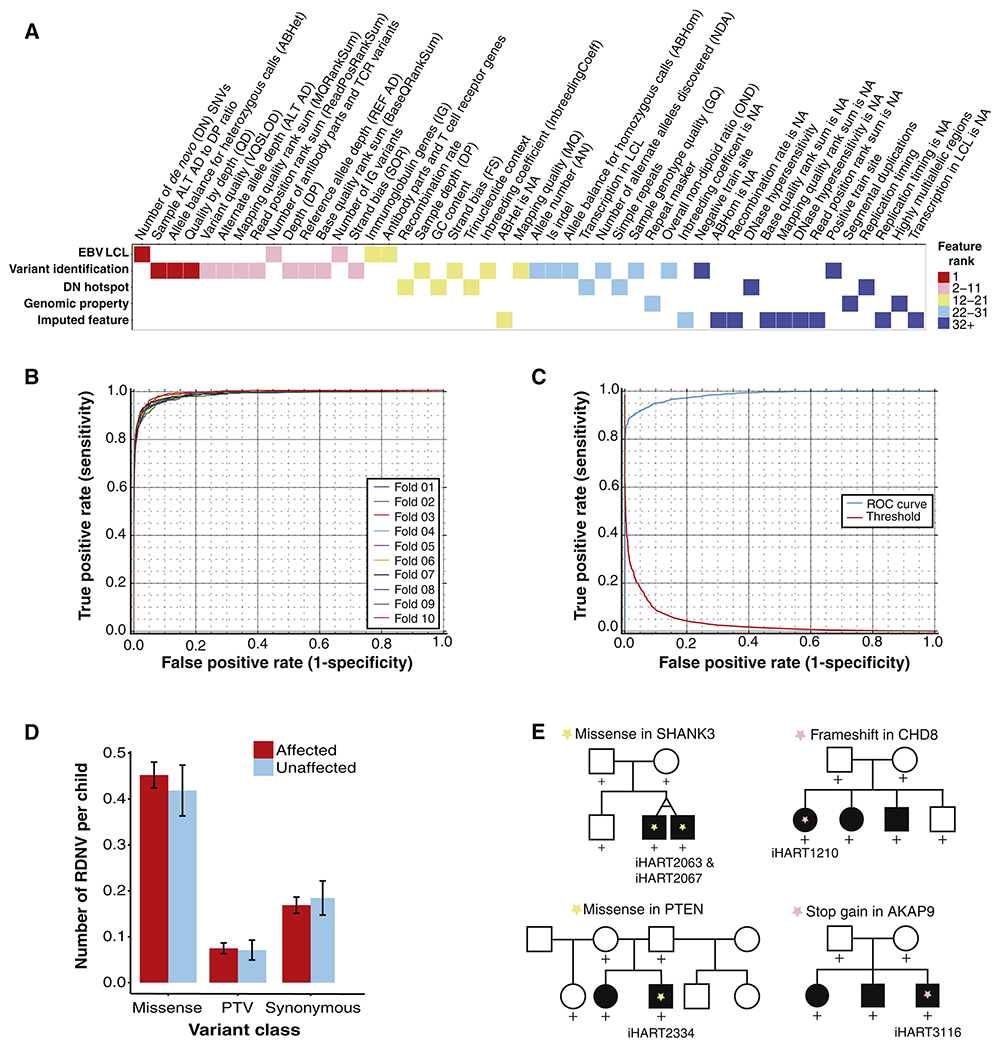
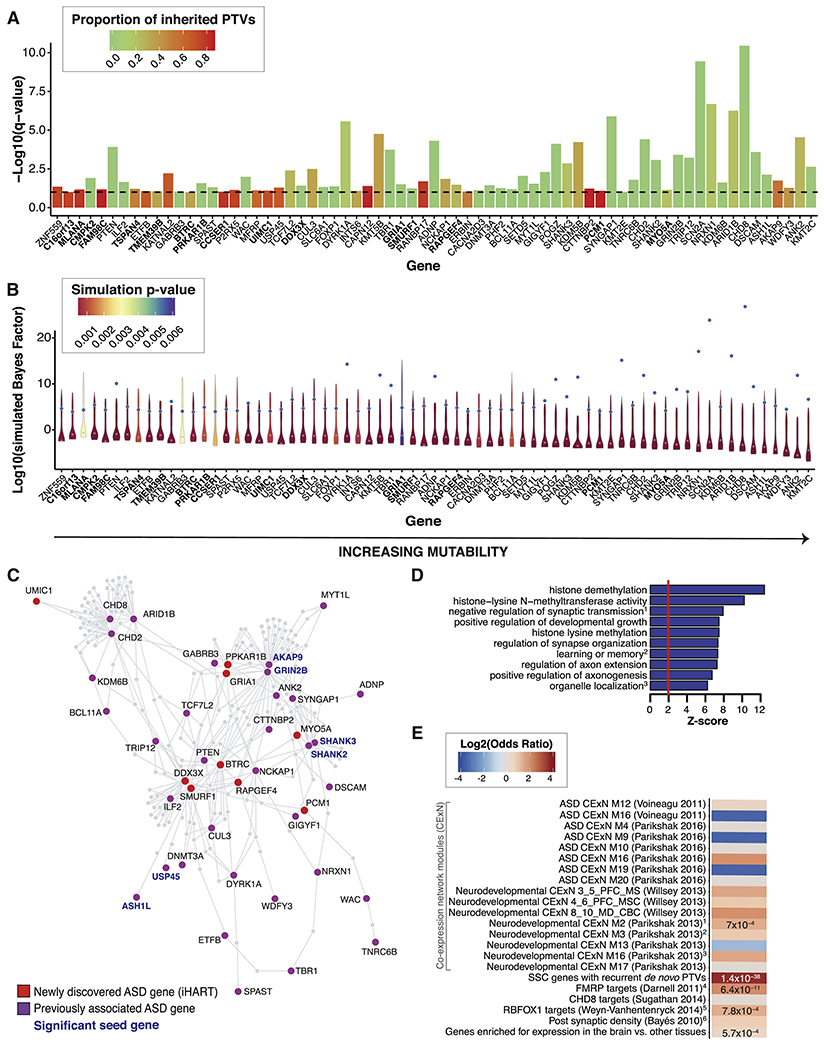
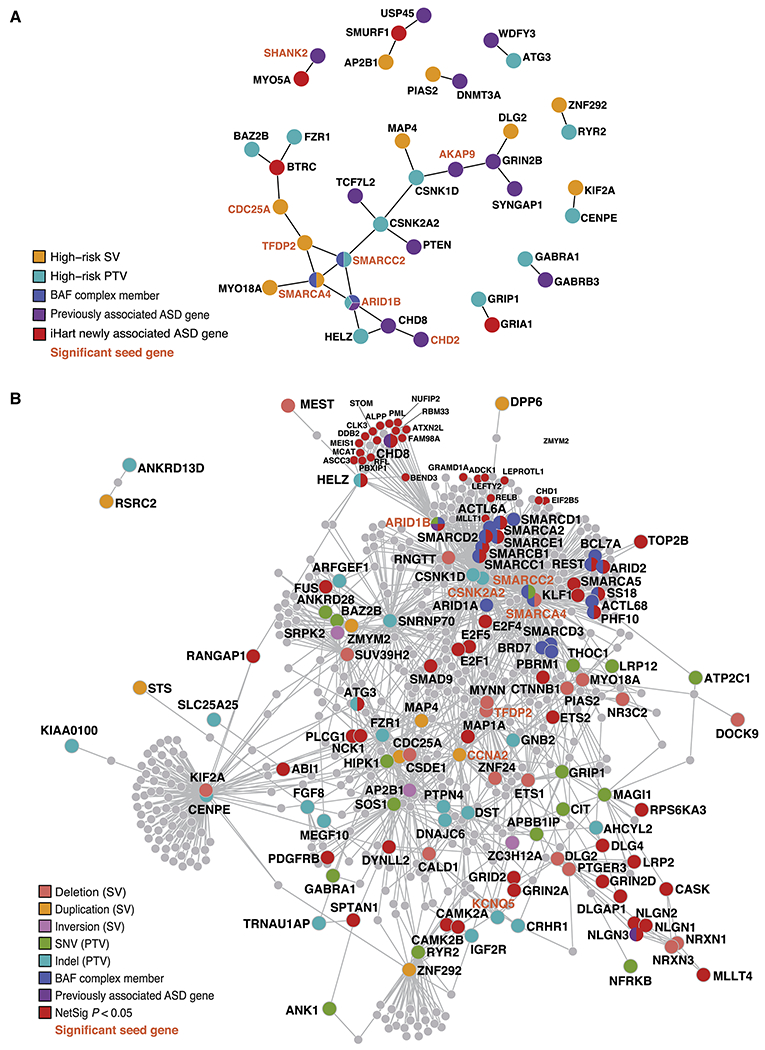
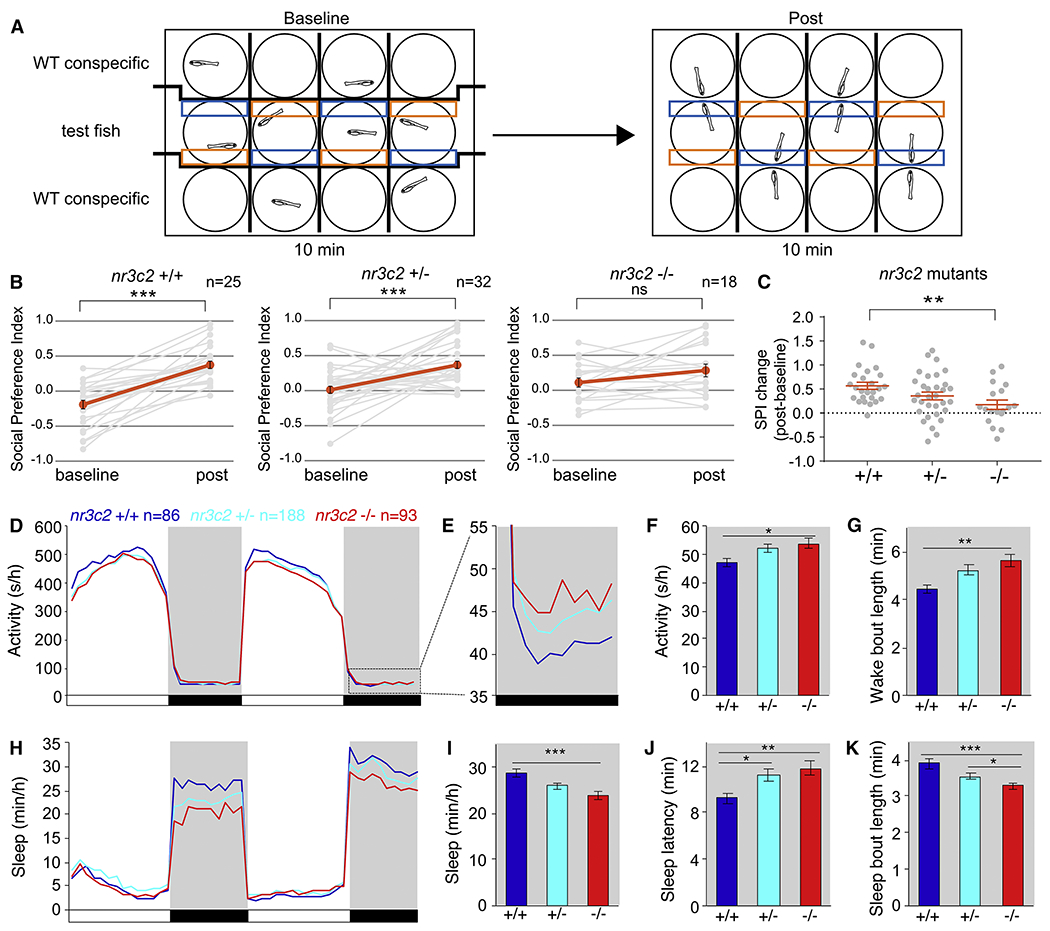
We performed a comprehensive assessment of rare inherited variation in autism spectrum disorder (ASD) by analyzing whole-genome sequences of 2,308 individuals from families with multiple affected children. We implicate 69 genes in ASD risk, including 24 passing genome-wide Bonferroni correction and 16 new ASD risk genes, most supported by rare inherited variants, a substantial extension of previous findings. Biological pathways enriched for genes harboring inherited variants represent cytoskeletal organization and ion transport, which are distinct from pathways implicated in previous studies. Nevertheless, the de novo and inherited genes contribute to a common protein-protein interaction network. We also identified structural variants (SVs) affecting non-coding regions, implicating recurrent deletions in the promoters of DLG2 and NR3C2. Loss of nr3c2 function in zebrafish disrupts sleep and social function, overlapping with human ASD-related phenotypes. These data support the utility of studying multiplex families in ASD and are available through the Hartwell Autism Research and Technology portal.
Keywords: ASD; autism; de novo; genetics; inherited; machine learning; multiplex families.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
References
-
- American Psychiatric Association; (2013). Diagnostic and Statistical Manual of Mental Disorders, 5th edn (Arlington, Virginia, USA: ).
-
- Bacchelli E, Blasi F, Biondolillo M, Lamb JA, Bonora E, Barnby G, Parr J, Beyer KS, Klauck SM, Poustka A, et al. (2003). Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Molecular psychiatry 8, 916–924. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
