

Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition
- PMID: 31399369
- PMCID: PMC7720933
- DOI: 10.1016/j.chom.2019.07.004
Obese Individuals with and without Type 2 Diabetes Show Different Gut Microbial Functional Capacity and Composition
Abstract
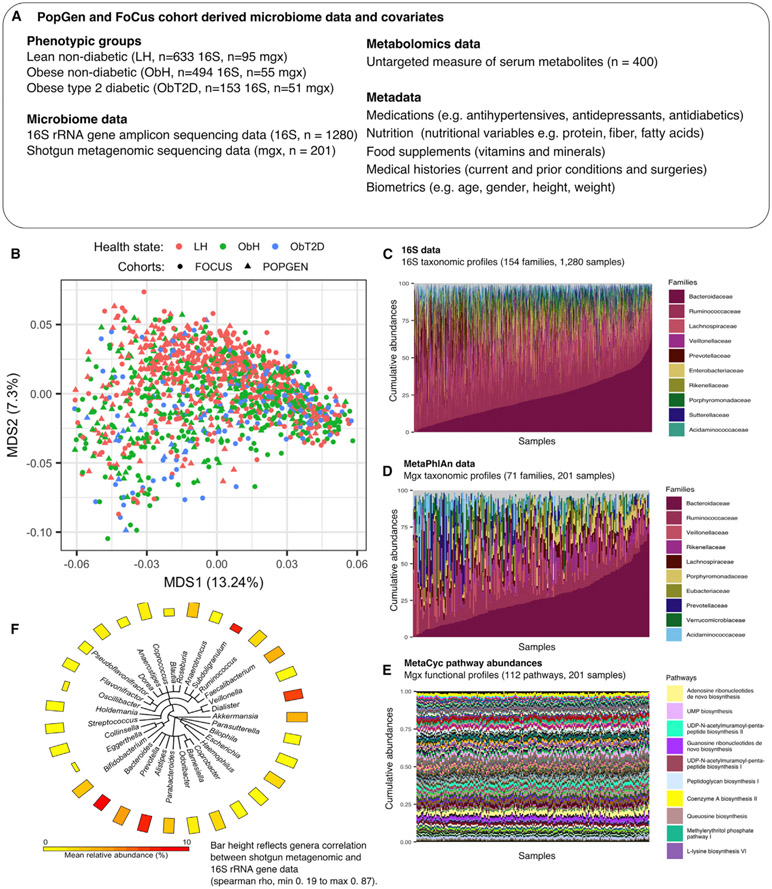
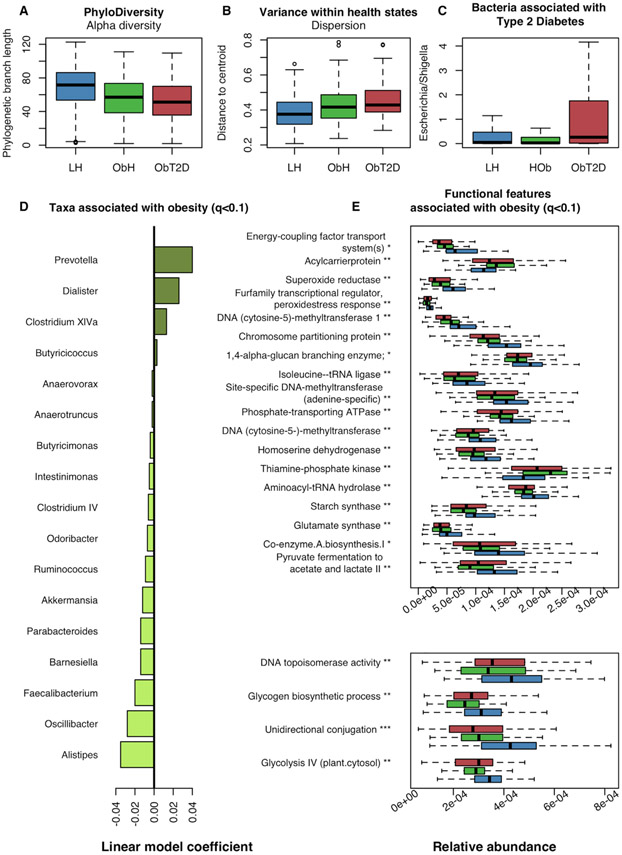
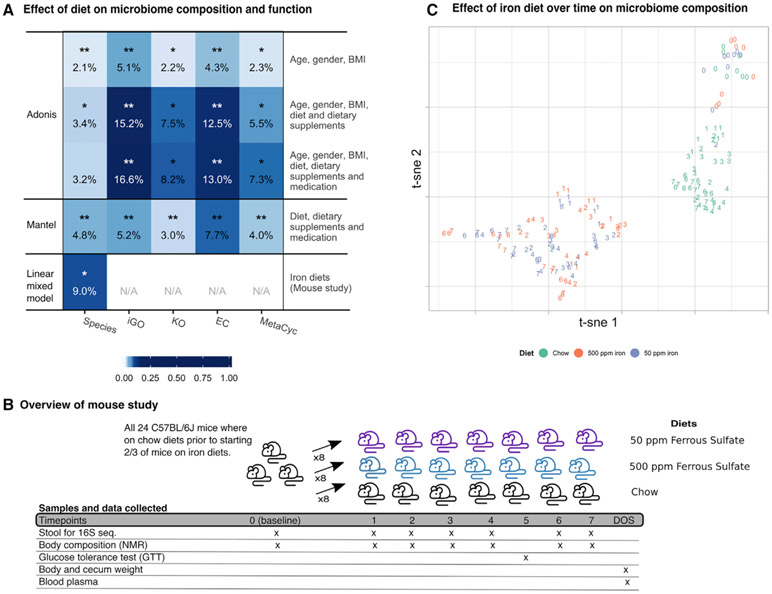
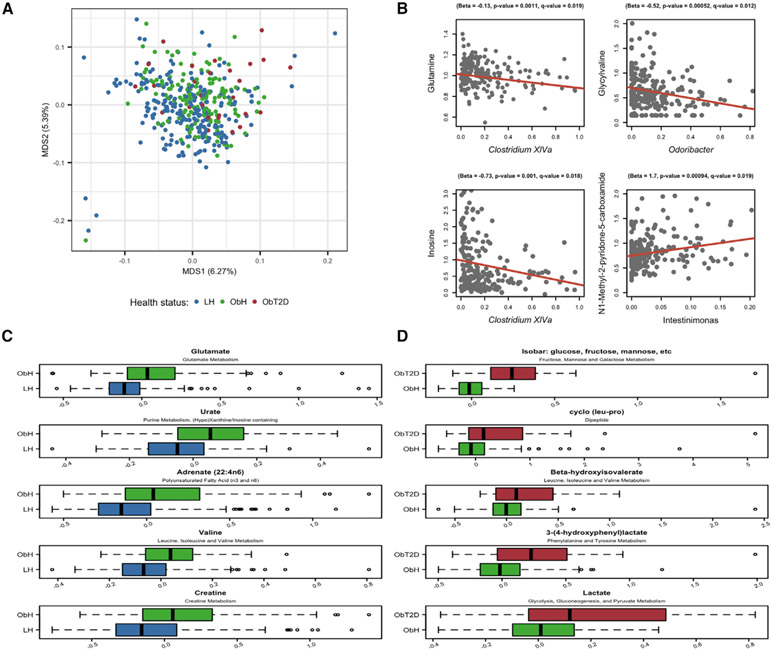
Obesity and type 2 diabetes (T2D) are metabolic disorders that are linked to microbiome alterations. However, their co-occurrence poses challenges in disentangling microbial features unique to each condition. We analyzed gut microbiomes of lean non-diabetic (n = 633), obese non-diabetic (n = 494), and obese individuals with T2D (n = 153) from German population and metabolic disease cohorts. Microbial taxonomic and functional profiles were analyzed along with medical histories, serum metabolomics, biometrics, and dietary data. Obesity was associated with alterations in microbiome composition, individual taxa, and functions with notable changes in Akkermansia, Faecalibacterium, Oscillibacter, and Alistipes, as well as in serum metabolites that correlated with gut microbial patterns. However, microbiome associations were modest for T2D, with nominal increases in Escherichia/Shigella. Medications, including antihypertensives and antidiabetics, along with dietary supplements including iron, were significantly associated with microbiome variation. These results differentiate microbial components of these interrelated metabolic diseases and identify dietary and medication exposures to consider in future studies.
Keywords: dietary supplements; iron; magnesium; medication; metabolic disease; microbiome; nutrition; obesity; type 2 diabetes.
Copyright © 2019. Published by Elsevier Inc.
Conflict of interest statement
DECLARATION OF INTERESTS
L.B.T. is an employee and shareholder of BiomCare. C.H. is a scientific advisor for Seres Therapeutics, microbiome Insights, and ZOE.
Figures
Comment in
-
Variations in microbial composition and function in metabolic diseases.Nat Rev Endocrinol. 2019 Oct;15(10):563. doi: 10.1038/s41574-019-0253-z. Nat Rev Endocrinol. 2019. PMID: 31417192 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
