

Targeting sickle cell disease root-cause pathophysiology with small molecules
- PMID: 31399526
- PMCID: PMC6717594
- DOI: 10.3324/haematol.2018.207530
Targeting sickle cell disease root-cause pathophysiology with small molecules
Abstract
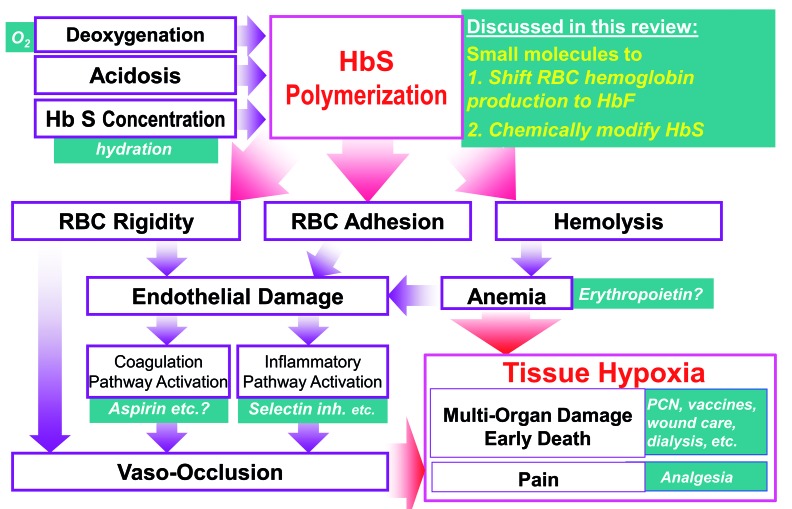
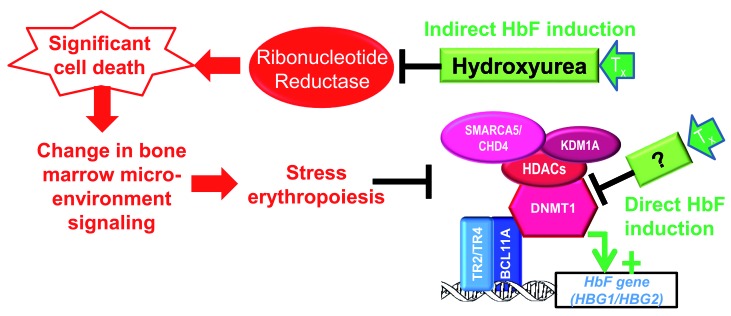
The complex, frequently devastating, multi-organ pathophysiology of sickle cell disease has a single root cause: polymerization of deoxygenated sickle hemoglobin. A logical approach to disease modification is, therefore, to interdict this root cause. Ideally, such interdiction would utilize small molecules that are practical and accessible for worldwide application. Two types of such small molecule strategies are actively being evaluated in the clinic. The first strategy intends to shift red blood cell precursor hemoglobin manufacturing away from sickle hemoglobin and towards fetal hemoglobin, which inhibits sickle hemoglobin polymerization by a number of mechanisms. The second strategy intends to chemically modify sickle hemoglobin directly in order to inhibit its polymerization. Important lessons have been learnt from the pre-clinical and clinical evaluations to date. Open questions remain, but this review summarizes the valuable experience and knowledge already gained, which can guide ongoing and future efforts for molecular mechanism-based, practical and accessible disease modification of sickle cell disease.
Copyright© 2019 Ferrata Storti Foundation.
Figures
References
-
- Saraf S, Farooqui M, Infusino G, et al. Standard clinical practice underestimates the role and significance of erythropoietin deficiency in sickle cell disease. Br J Haematol. 2011;153(3):386–392. - PubMed
-
- Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG. The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009;84(6):323–327. - PubMed
-
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
