

Distance-based protein folding powered by deep learning
- PMID: 31399549
- PMCID: PMC6708335
- DOI: 10.1073/pnas.1821309116
Distance-based protein folding powered by deep learning
Abstract
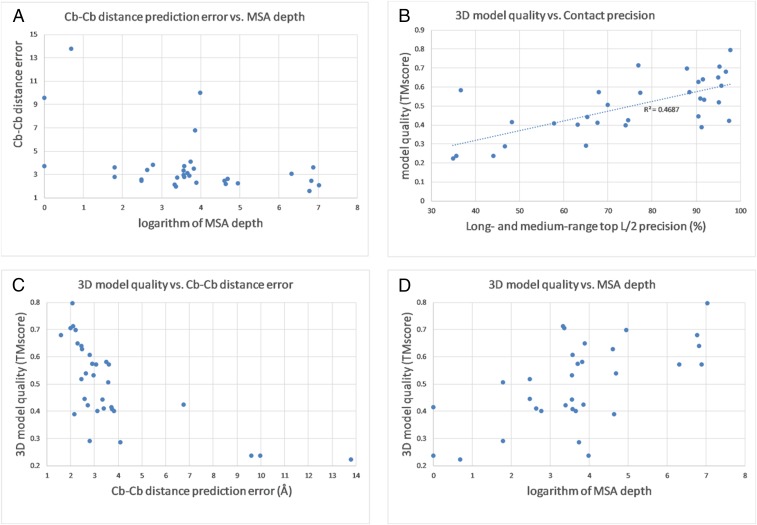
Direct coupling analysis (DCA) for protein folding has made very good progress, but it is not effective for proteins that lack many sequence homologs, even coupled with time-consuming conformation sampling with fragments. We show that we can accurately predict interresidue distance distribution of a protein by deep learning, even for proteins with ∼60 sequence homologs. Using only the geometric constraints given by the resulting distance matrix we may construct 3D models without involving extensive conformation sampling. Our method successfully folded 21 of the 37 CASP12 hard targets with a median family size of 58 effective sequence homologs within 4 h on a Linux computer of 20 central processing units. In contrast, DCA-predicted contacts cannot be used to fold any of these hard targets in the absence of extensive conformation sampling, and the best CASP12 group folded only 11 of them by integrating DCA-predicted contacts into fragment-based conformation sampling. Rigorous experimental validation in CASP13 shows that our distance-based folding server successfully folded 17 of 32 hard targets (with a median family size of 36 sequence homologs) and obtained 70% precision on the top L/5 long-range predicted contacts. The latest experimental validation in CAMEO shows that our server predicted correct folds for 2 membrane proteins while all of the other servers failed. These results demonstrate that it is now feasible to predict correct fold for many more proteins lack of similar structures in the Protein Data Bank even on a personal computer.
Keywords: deep learning; direct coupling analysis; protein contact prediction; protein distance prediction; protein folding.
Conflict of interest statement
The author declares no conflict of interest.
Figures





References
-
- de Juan D., Pazos F., Valencia A., Emerging methods in protein co-evolution. Nat. Rev. Genet. 14, 249–261 (2013). - PubMed
-
- Jones D. T., Buchan D. W., Cozzetto D., Pontil M., PSICOV: Precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics 28, 184–190 (2012). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
