

Extreme Polygenicity of Complex Traits Is Explained by Negative Selection
- PMID: 31402091
- PMCID: PMC6732528
- DOI: 10.1016/j.ajhg.2019.07.003
Extreme Polygenicity of Complex Traits Is Explained by Negative Selection
Abstract
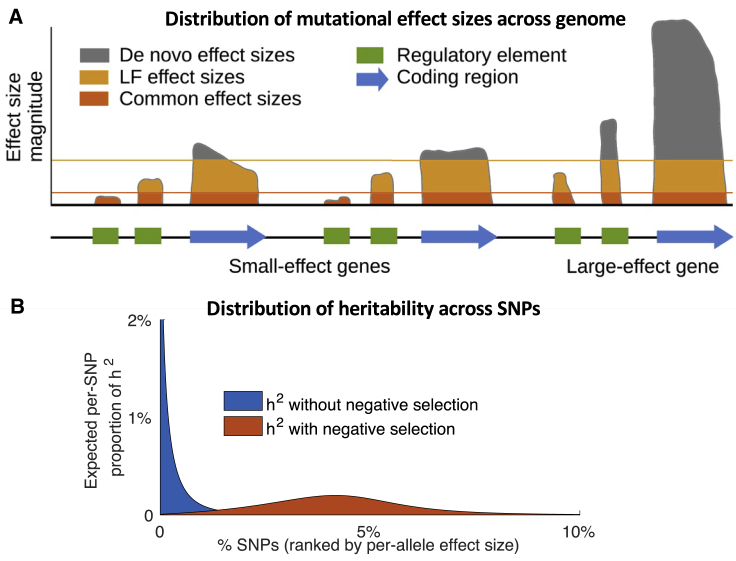
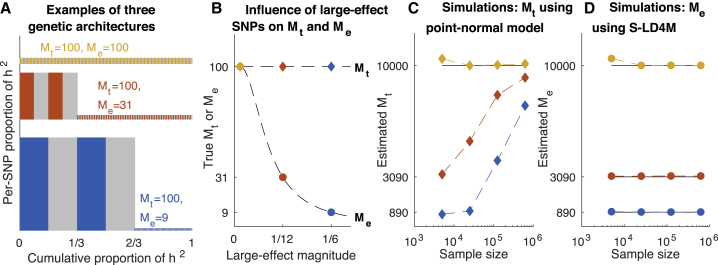
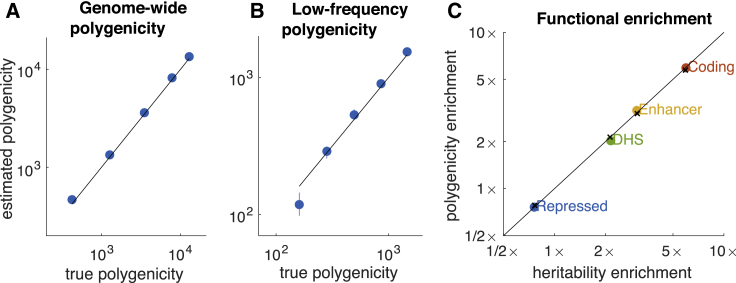
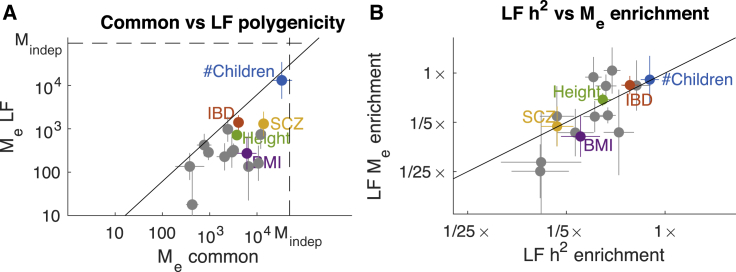
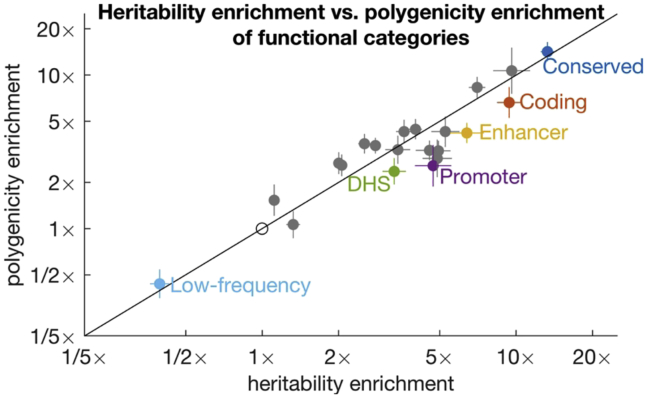
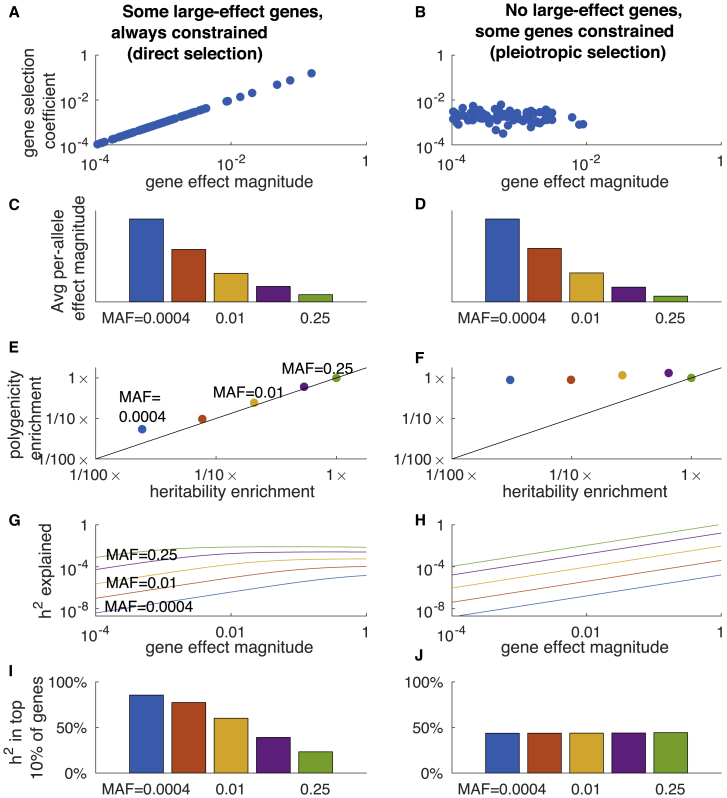
Complex traits and common diseases are extremely polygenic, their heritability spread across thousands of loci. One possible explanation is that thousands of genes and loci have similarly important biological effects when mutated. However, we hypothesize that for most complex traits, relatively few genes and loci are critical, and negative selection-purging large-effect mutations in these regions-leaves behind common-variant associations in thousands of less critical regions instead. We refer to this phenomenon as flattening. To quantify its effects, we introduce a mathematical definition of polygenicity, the effective number of independently associated SNPs (Me), which describes how evenly the heritability of a trait is spread across the genome. We developed a method, stratified LD fourth moments regression (S-LD4M), to estimate Me, validating that it produces robust estimates in simulations. Analyzing 33 complex traits (average N = 361k), we determined that heritability is spread ∼4× more evenly among common SNPs than among low-frequency SNPs. This difference, together with evolutionary modeling of new mutations, suggests that complex traits would be orders of magnitude less polygenic if not for the influence of negative selection. We also determined that heritability is spread more evenly within functionally important regions in proportion to their heritability enrichment; functionally important regions do not harbor common SNPs with greatly increased causal effect sizes, due to selective constraint. Our results suggest that for most complex traits, the genes and loci with the most critical biological effects often differ from those with the strongest common-variant associations.
Keywords: GWAS; SLD4M; heritability; negative selection; polygenicity.
Copyright © 2019 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Stahl E.A., Wegmann D., Trynka G., Gutierrez-Achury J., Do R., Voight B.F., Kraft P., Chen R., Kallberg H.J., Kurreeman F.A., Diabetes Genetics Replication and Meta-analysis Consortium. Myocardial Infarction Genetics Consortium Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat. Genet. 2012;44:483–489. - PMC - PubMed
-
- Loh P.R., Bhatia G., Gusev A., Finucane H.K., Bulik-Sullivan B.K., Pollack S.J., de Candia T.R., Lee S.H., Wray N.R., Kendler K.S., Schizophrenia Working Group of Psychiatric Genomics Consortium Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat. Genet. 2015;47:1385–1392. - PMC - PubMed
-
- Bulik-Sullivan B.K., Loh P.-R., Finucane H.K., Ripke S., Yang J., Patterson N., Daly M.J., Price A.L., Neale B.M., Schizophrenia Working Group of the Psychiatric Genomics Consortium LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 2015;47:291–295. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
