

MARPLE, a point-of-care, strain-level disease diagnostics and surveillance tool for complex fungal pathogens
- PMID: 31405370
- PMCID: PMC6691556
- DOI: 10.1186/s12915-019-0684-y
MARPLE, a point-of-care, strain-level disease diagnostics and surveillance tool for complex fungal pathogens
Abstract
Background: Effective disease management depends on timely and accurate diagnosis to guide control measures. The capacity to distinguish between individuals in a pathogen population with specific properties such as fungicide resistance, toxin production and virulence profiles is often essential to inform disease management approaches. The genomics revolution has led to technologies that can rapidly produce high-resolution genotypic information to define individual variants of a pathogen species. However, their application to complex fungal pathogens has remained limited due to the frequent inability to culture these pathogens in the absence of their host and their large genome sizes.
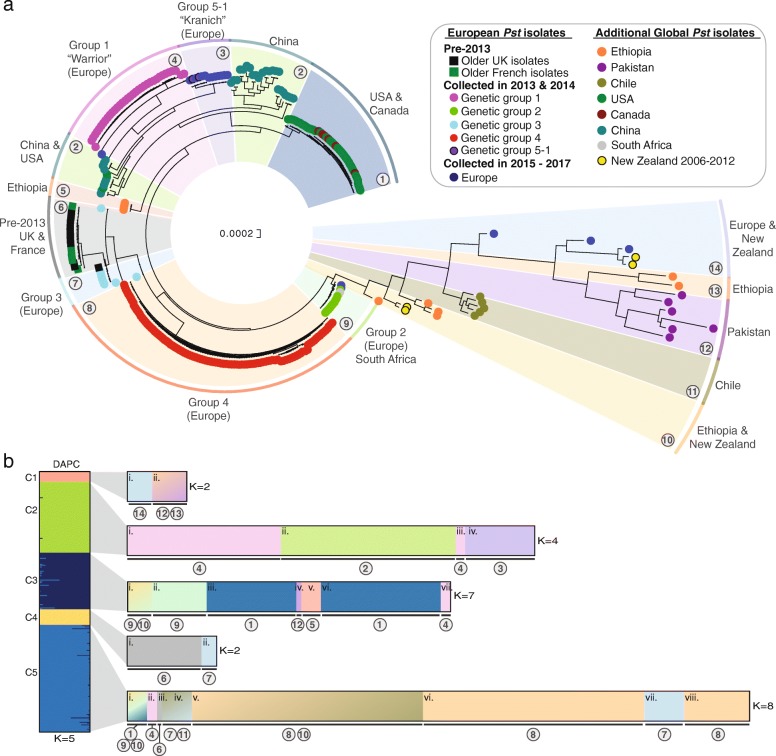
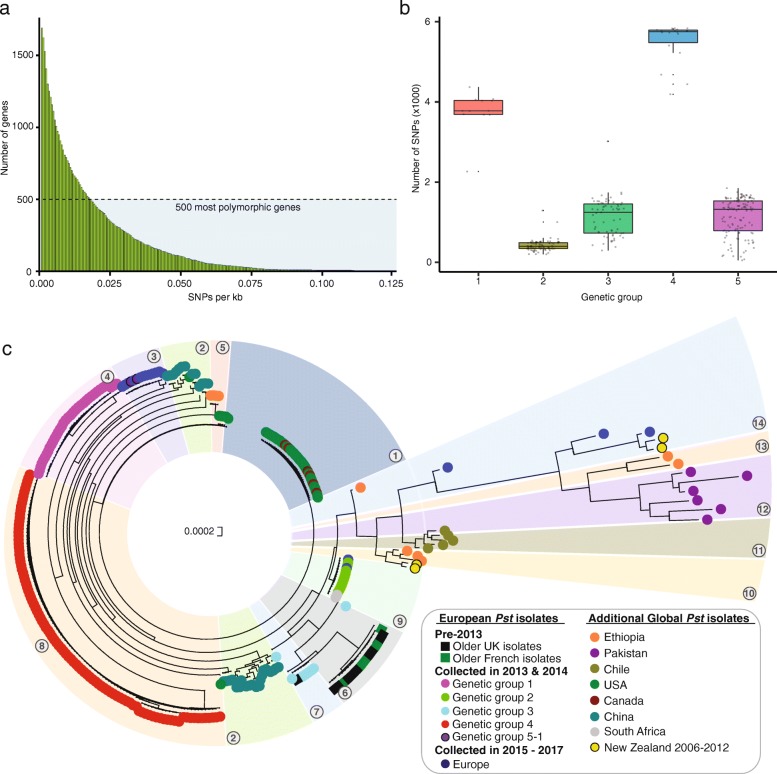
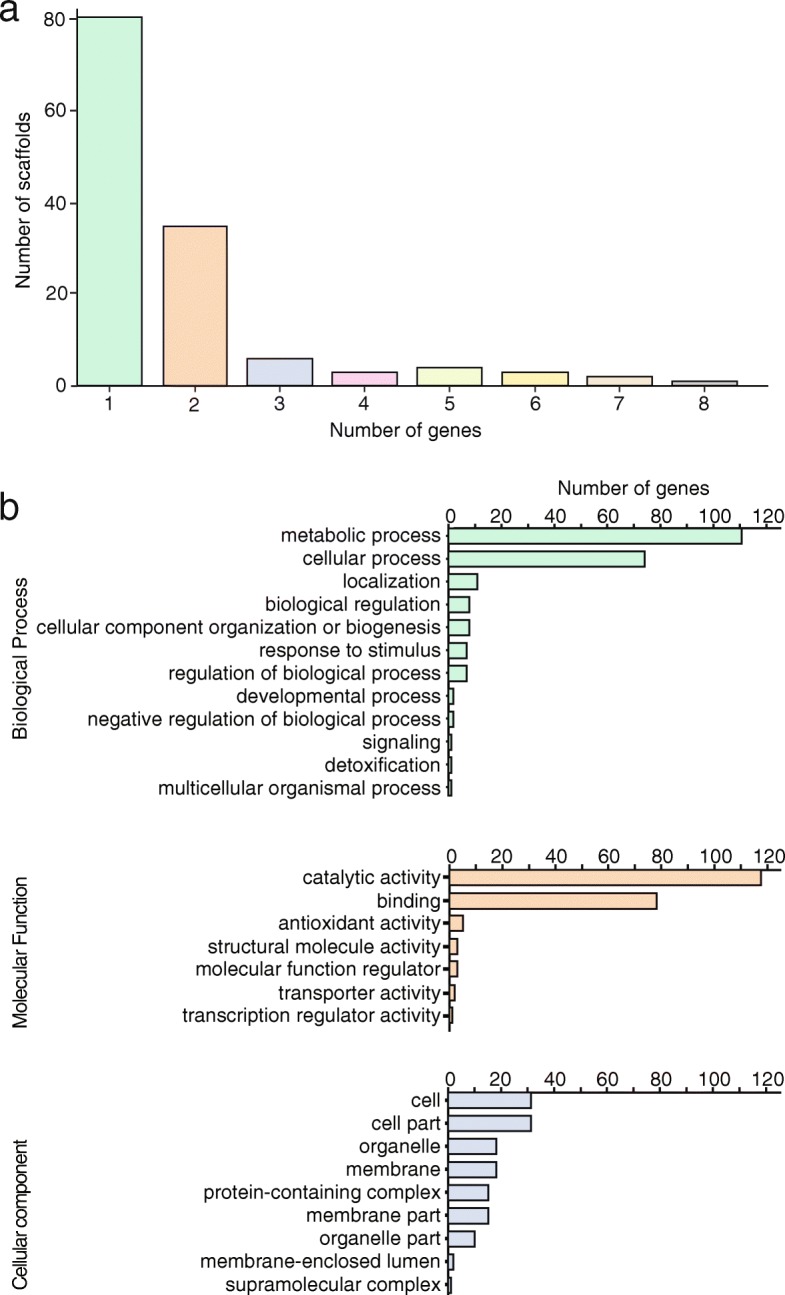
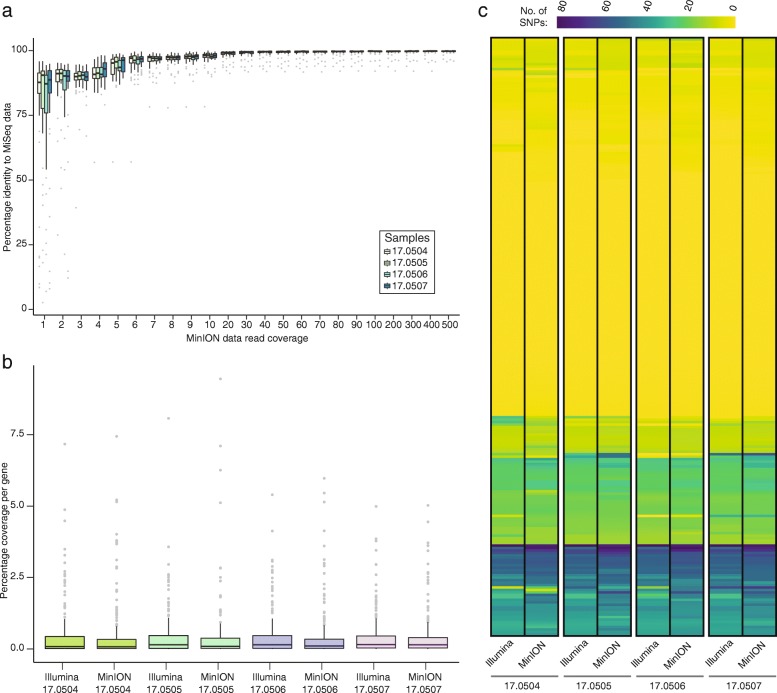
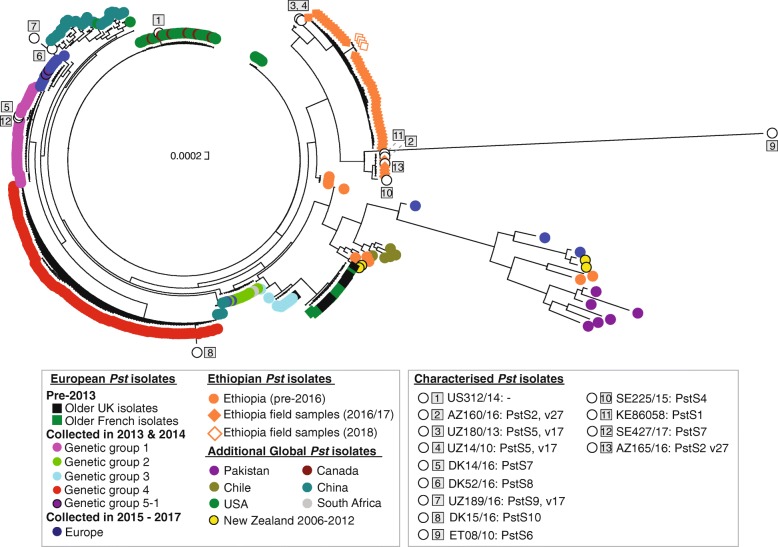
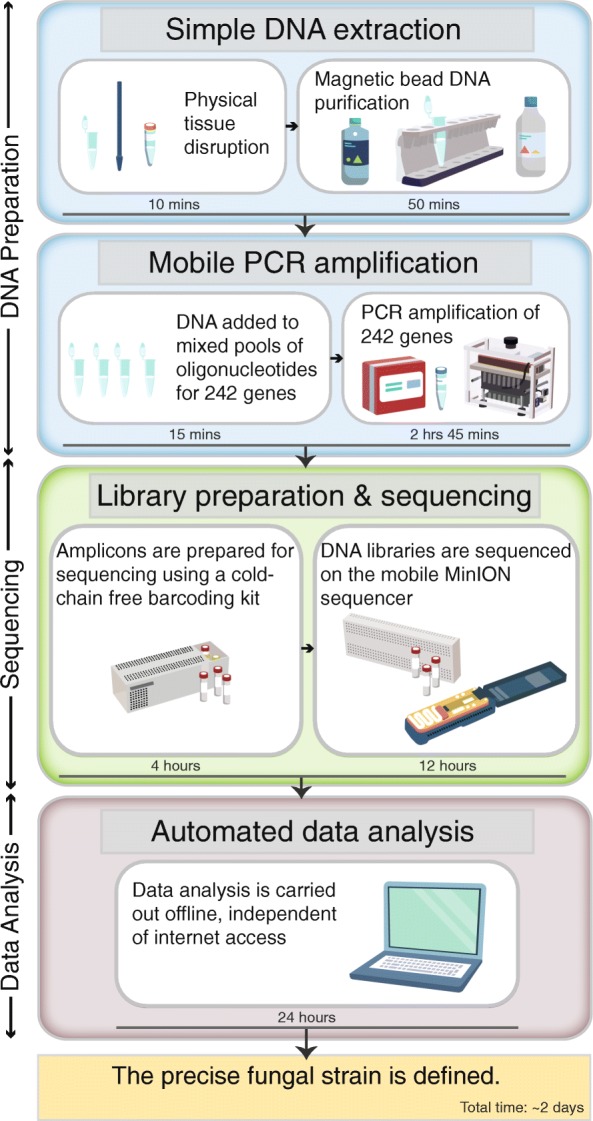
Results: Here, we describe the development of Mobile And Real-time PLant disEase (MARPLE) diagnostics, a portable, genomics-based, point-of-care approach specifically tailored to identify individual strains of complex fungal plant pathogens. We used targeted sequencing to overcome limitations associated with the size of fungal genomes and their often obligately biotrophic nature. Focusing on the wheat yellow rust pathogen, Puccinia striiformis f.sp. tritici (Pst), we demonstrate that our approach can be used to rapidly define individual strains, assign strains to distinct genetic lineages that have been shown to correlate tightly with their virulence profiles and monitor genes of importance.
Conclusions: MARPLE diagnostics enables rapid identification of individual pathogen strains and has the potential to monitor those with specific properties such as fungicide resistance directly from field-collected infected plant tissue in situ. Generating results within 48 h of field sampling, this new strategy has far-reaching implications for tracking plant health threats.
Keywords: Disease diagnostics; Genomics; Nanopore sequencing; Pathogen surveillance; Point of care; Wheat rust.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Martinelli F, Scalenghe R, Davino S, Panno S, Scuderi G, Ruisi P, et al. Advanced methods of plant disease detection. A review. Agron Sustain Dev. 2015;35(1):1–25. doi: 10.1007/s13593-014-0246-1. - DOI
-
- Ward E, Foster SJ, Fraaije BA, McCartney HA. Plant pathogen diagnostics: immunological and nucleic acid-based approaches. Ann Appl Biol. 2004;145(1):1–16. doi: 10.1111/j.1744-7348.2004.tb00354.x. - DOI
-
- Van Vuurde JWL. New approach in detecting phytopathogenic bacteria by combined immunoisolation and immunoidentification assays. EPPO Bulletin. 1987;17(1):139–148. doi: 10.1111/j.1365-2338.1987.tb00019.x. - DOI
Publication types
MeSH terms
Grants and funding
- BB/J004553/1 and BB/P012574/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/GCRF-IAA/11 and BB/GCRF-IAA/17/11/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/N503964/2/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BBS/OS/GC/000013A/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- BB/M011216/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Medical
