

Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1
- PMID: 31409373
- PMCID: PMC6693144
- DOI: 10.1186/s13059-019-1753-9
Screening for genes that accelerate the epigenetic aging clock in humans reveals a role for the H3K36 methyltransferase NSD1
Abstract
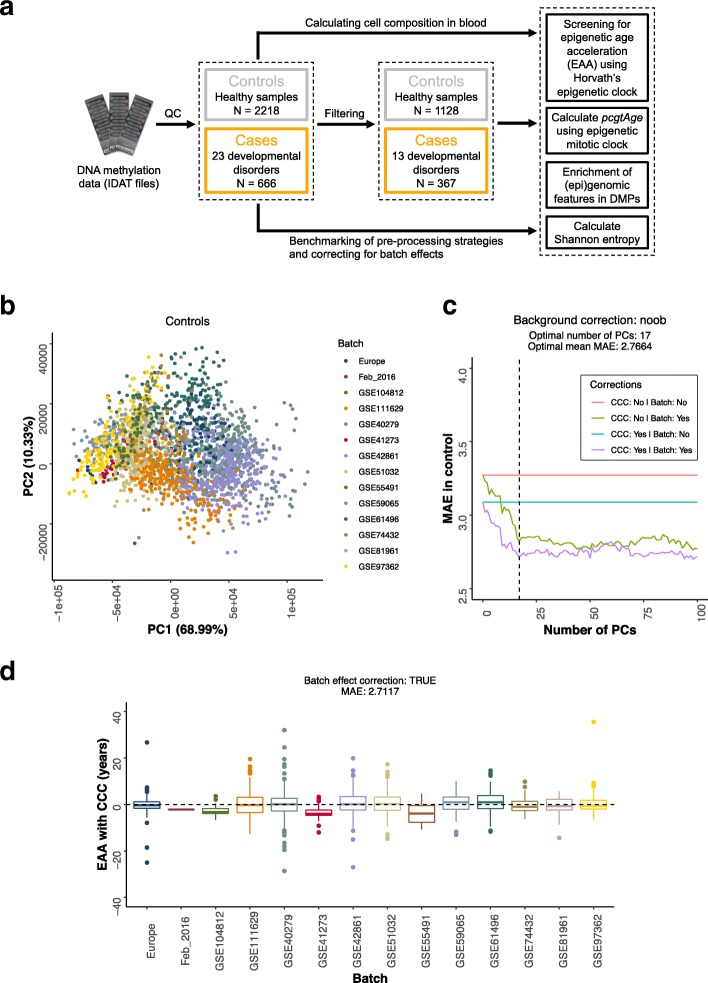
Background: Epigenetic clocks are mathematical models that predict the biological age of an individual using DNA methylation data and have emerged in the last few years as the most accurate biomarkers of the aging process. However, little is known about the molecular mechanisms that control the rate of such clocks. Here, we have examined the human epigenetic clock in patients with a variety of developmental disorders, harboring mutations in proteins of the epigenetic machinery.
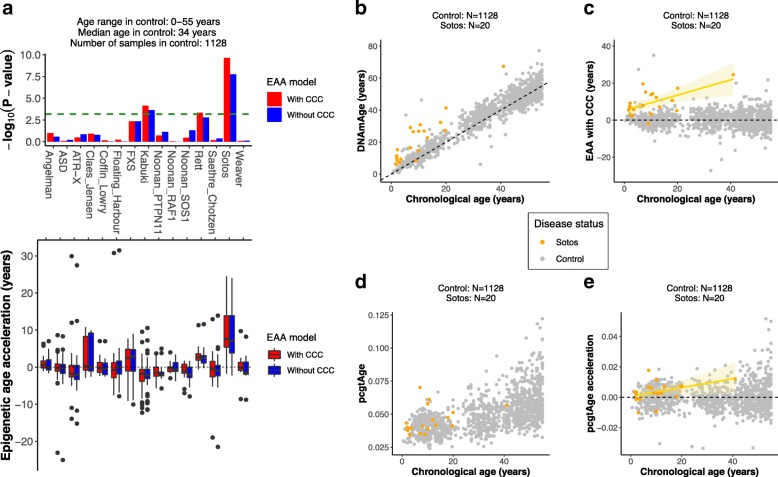
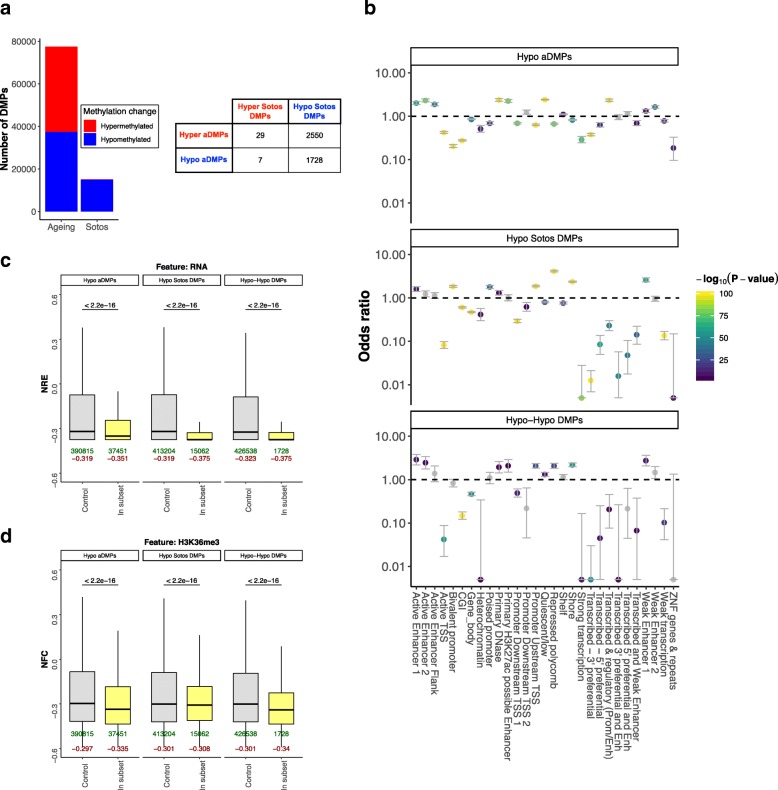
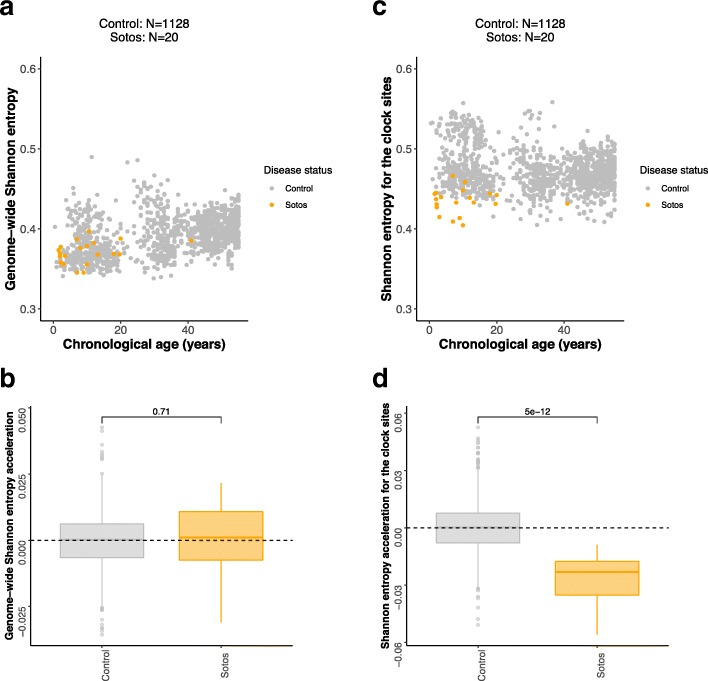
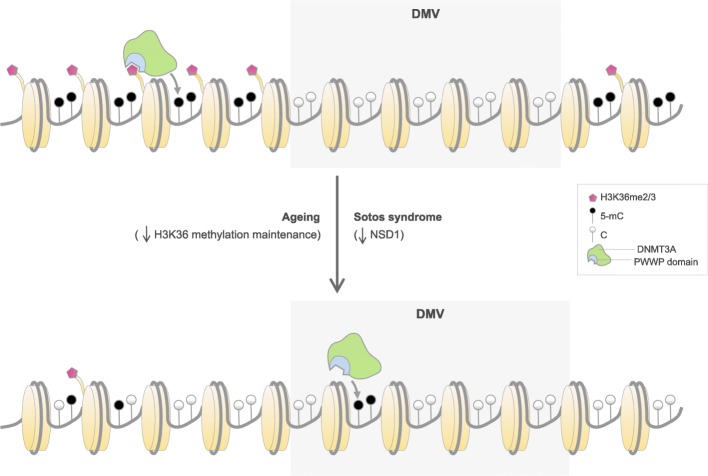
Results: Using the Horvath epigenetic clock, we perform an unbiased screen for epigenetic age acceleration in the blood of these patients. We demonstrate that loss-of-function mutations in the H3K36 histone methyltransferase NSD1, which cause Sotos syndrome, substantially accelerate epigenetic aging. Furthermore, we show that the normal aging process and Sotos syndrome share methylation changes and the genomic context in which they occur. Finally, we found that the Horvath clock CpG sites are characterized by a higher Shannon methylation entropy when compared with the rest of the genome, which is dramatically decreased in Sotos syndrome patients.
Conclusions: These results suggest that the H3K36 methylation machinery is a key component of the epigenetic maintenance system in humans, which controls the rate of epigenetic aging, and this role seems to be conserved in model organisms. Our observations provide novel insights into the mechanisms behind the epigenetic aging clock and we expect will shed light on the different processes that erode the human epigenetic landscape during aging.
Keywords: Aging; Biological age; DNA methylation; Developmental disorder; Epigenetic clock; Epigenetics; H3K36 methylation; Methylation entropy; NSD1; Sotos syndrome.
Conflict of interest statement
DEMH and TMS are founders and shareholders of Chronomics Limited, a UK-based company that provides epigenetic testing. WR is a consultant and shareholder of Cambridge Epigenetix. All other authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
