

Master transcription factors form interconnected circuitry and orchestrate transcriptional networks in oesophageal adenocarcinoma
- PMID: 31409603
- PMCID: PMC8108390
- DOI: 10.1136/gutjnl-2019-318325
Master transcription factors form interconnected circuitry and orchestrate transcriptional networks in oesophageal adenocarcinoma
Abstract
Objective: While oesophageal squamous cell carcinoma remains infrequent in Western populations, the incidence of oesophageal adenocarcinoma (EAC) has increased sixfold to eightfold over the past four decades. We aimed to characterise oesophageal cancer-specific and subtypes-specific gene regulation patterns and their upstream transcription factors (TFs). DESIGN: To identify regulatory elements, we profiled fresh-frozen oesophageal normal samples, tumours and cell lines with chromatin immunoprecipitation sequencing (ChIP-Seq). Mathematical modelling was performed to establish (super)-enhancers landscapes and interconnected transcriptional circuitry formed by master TFs. Coregulation and cooperation between master TFs were investigated by ChIP-Seq, circularised chromosome conformation capture sequencing and luciferase assay. Biological functions of candidate factors were evaluated both in vitro and in vivo.
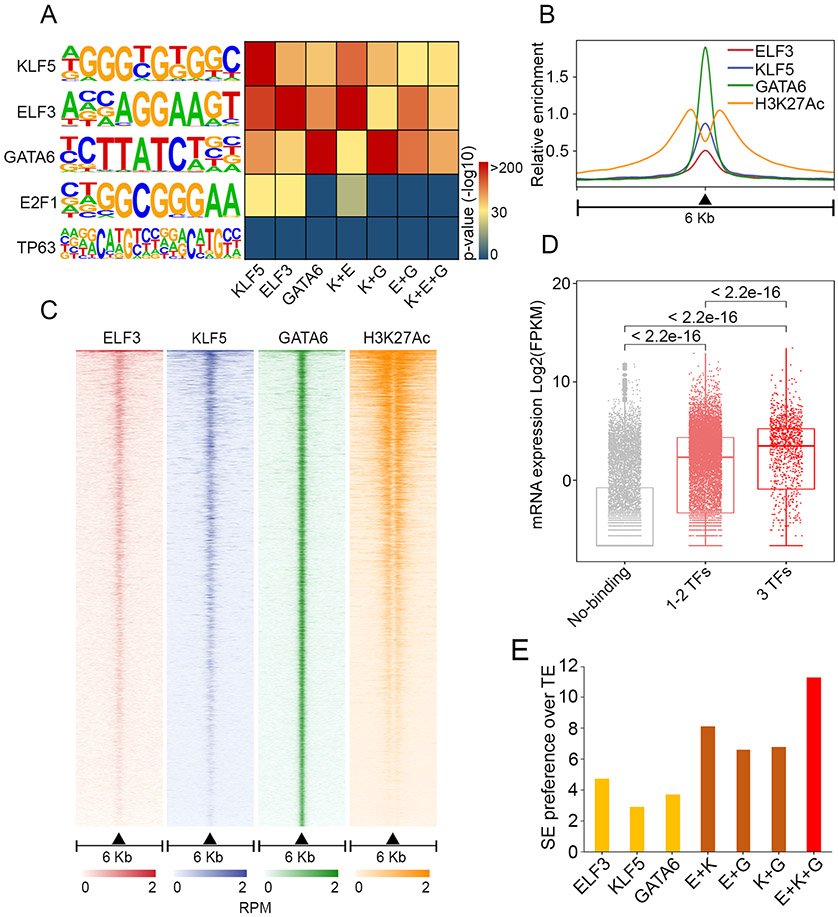
Results: We found widespread and pervasive alterations of the (super)-enhancer reservoir in both subtypes of oesophageal cancer, leading to transcriptional activation of a myriad of novel oncogenes and signalling pathways, some of which may be exploited pharmacologically (eg, leukemia inhibitory factor (LIF) pathway). Focusing on EAC, we bioinformatically reconstructed and functionally validated an interconnected circuitry formed by four master TFs-ELF3, KLF5, GATA6 and EHF-which promoted each other's expression by interacting with each super-enhancer. Downstream, these master TFs occupied almost all EAC super-enhancers and cooperatively orchestrated EAC transcriptome. Each TF within the transcriptional circuitry was highly and specifically expressed in EAC and functionally promoted EAC cell proliferation and survival.
Conclusions: By establishing cancer-specific and subtype-specific features of the EAC epigenome, our findings promise to transform understanding of the transcriptional dysregulation and addiction of EAC, while providing molecular clues to develop novel therapeutic modalities against this malignancy.
Keywords: gene regulation; oesophageal cancer; signal transduction; transcription factor.
© Author(s) (or their employer(s)) 2020. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures





References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous