

Lumacaftor-ivacaftor in the treatment of cystic fibrosis: design, development and place in therapy
- PMID: 31409974
- PMCID: PMC6650604
- DOI: 10.2147/DDDT.S153719
Lumacaftor-ivacaftor in the treatment of cystic fibrosis: design, development and place in therapy
Abstract
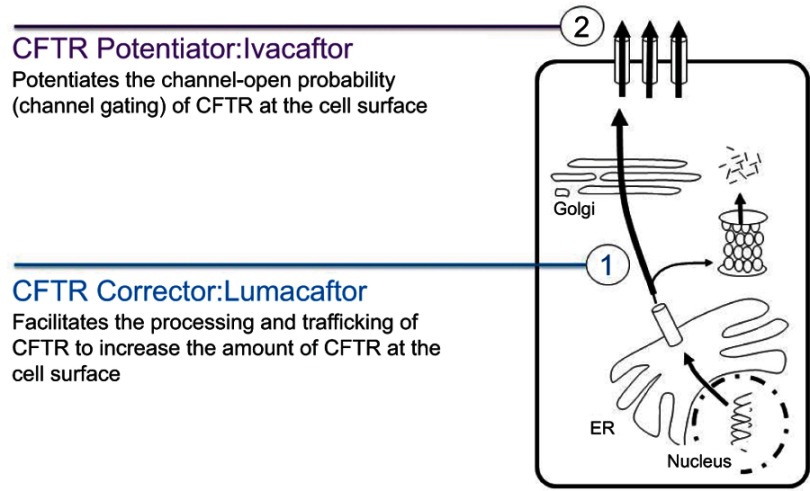
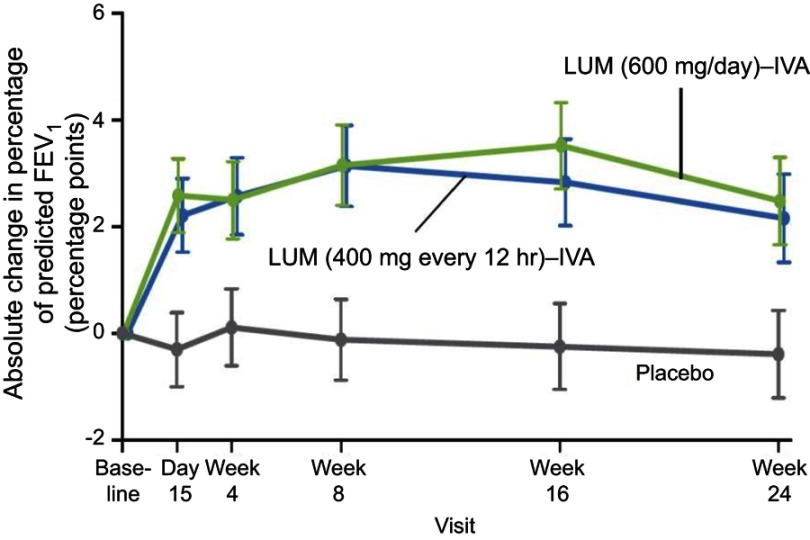
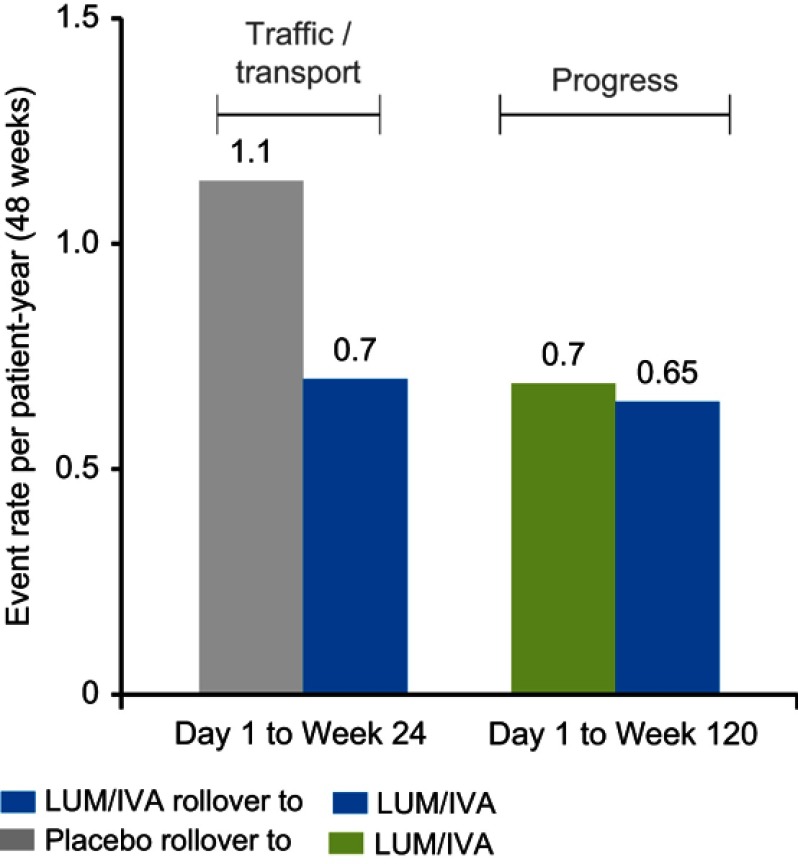
Lumacaftor-ivacaftor is a combination of two small molecule therapies targeting the basic defect in cystic fibrosis (CF) at a cellular level. It is a precision medicine and its effects are specific to individuals with two copies of the p.Phe508del gene mutation. The drug combination works by restoring functioning CF transmembrane conductance regulator (CFTR) protein in cell surface membranes and was the first CFTR modulator licensed for the homozygous p.Phe508del genotype. The drug is a combination of a CFTR corrector and potentiator. Lumacaftor, the corrector, works by increasing the trafficking of CFTR proteins to the outer cell membrane. Ivacaftor, the potentiator, works by enabling the opening of what would otherwise be a dysfunctional chloride channel. In vivo lumacaftor-ivacaftor improves Phe508del-CFTR activity in airways, sweat ducts and intestine to approximately 10-20% of normal CFTR function with greater reductions in sweat chloride levels in children versus adults. Its use results in a modest improvement in lung function and a decreased rate of subsequent decline. Perhaps more importantly, those treated report increased levels of well-being and their rate of respiratory exacerbations is significantly improved. This review traces the development and use of this combination of CFTR modulators, the first licensed drug for treating the homozygous p.Phe508del CF genotype at the intracellular level by correcting the protein defect.
Keywords: DF508; Orkambi; Phe508del; corrector; modifier; modulator.
Conflict of interest statement
GJ Connett has been an investigator in Vertex, Flateley, Proteostasis, Zambon, Mylan and Corbus-sponsored cystic fibrosis clinical trials. The Southampton Children’s Hospital has received support for educational meetings from Vertex, Chiesi, Mylan, Novartis, PARI GmbH and Teva pharmaceuticals. GJ Connett reports personal fees from Vertex during the conduct of the study. The author reports no other conflicts of interest in this work.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
