

Congenital hyperinsulinism disorders: Genetic and clinical characteristics
- PMID: 31414570
- PMCID: PMC7229866
- DOI: 10.1002/ajmg.c.31737
Congenital hyperinsulinism disorders: Genetic and clinical characteristics
Abstract
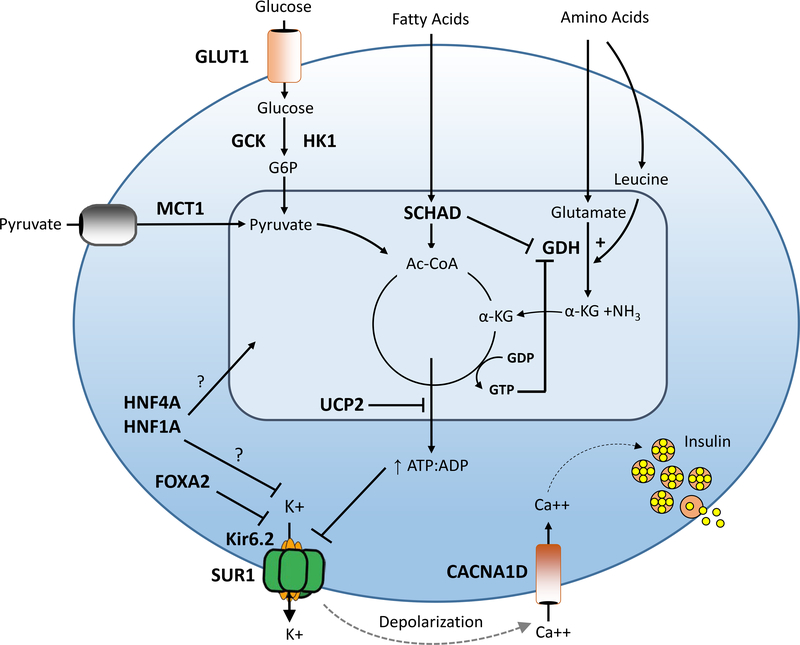
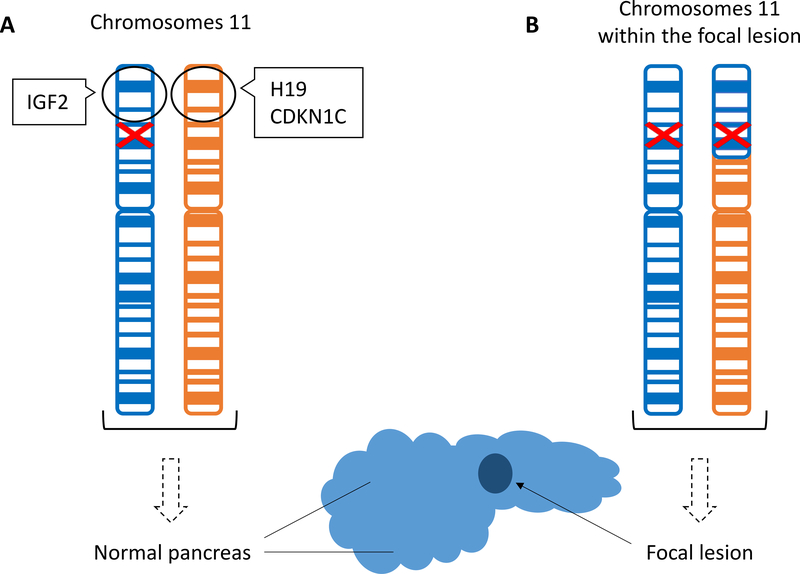
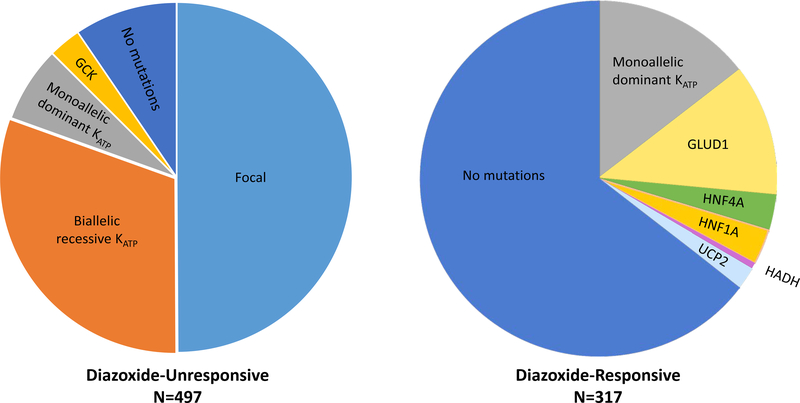
Congenital hyperinsulinism (HI) is the most frequent cause of persistent hypoglycemia in infants and children. Delays in diagnosis and initiation of appropriate treatment contribute to a high risk of neurocognitive impairment. HI represents a heterogeneous group of disorders characterized by dysregulated insulin secretion by the pancreatic beta cells, which in utero, may result in somatic overgrowth. There are at least nine known monogenic forms of HI as well as several syndromic forms. Molecular diagnosis allows for prediction of responsiveness to medical treatment and likelihood of surgically-curable focal hyperinsulinism. Timely genetic mutation analysis has thus become standard of care. However, despite significant advances in our understanding of the molecular basis of this disorder, the number of patients without an identified genetic diagnosis remains high, suggesting that there are likely additional genetic loci that have yet to be discovered.
Keywords: KATP channel; beta-cell; hypoglycemia; insulin; pancreas.
© 2019 Wiley Periodicals, Inc.
Figures
References
-
- Al Mutair AN, Brusgaard K, Bin-Abbas B, Hussain K, Felimban N, Al Shaikh A, & Christesen HT (2013). Heterogeneity in phenotype of usher-congenital hyperinsulinism syndrome: hearing loss, retinitis pigmentosa, and hyperinsulinemic hypoglycemia ranging from severe to mild with conversion to diabetes. Diabetes Care, 36(3), 557–561. doi: 10.2337/dc12-1174 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
