

The Landscape of Genetic Content in the Gut and Oral Human Microbiome
- PMID: 31415755
- PMCID: PMC6716383
- DOI: 10.1016/j.chom.2019.07.008
The Landscape of Genetic Content in the Gut and Oral Human Microbiome
Abstract
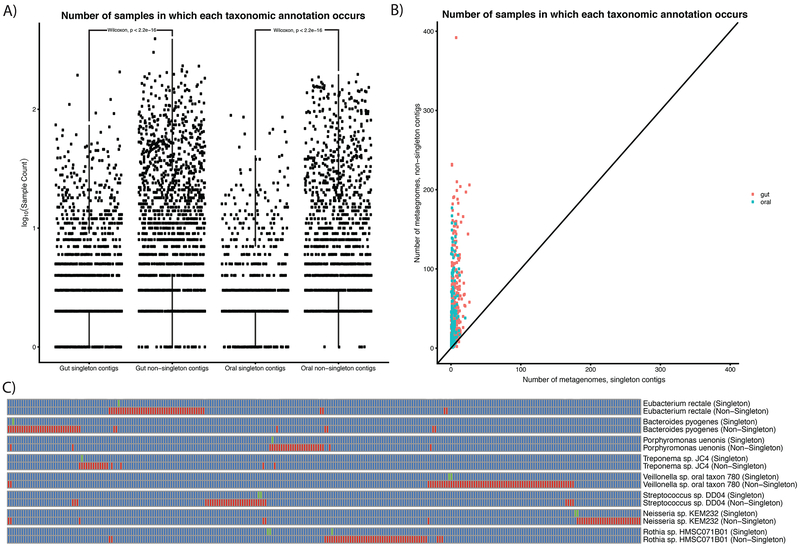
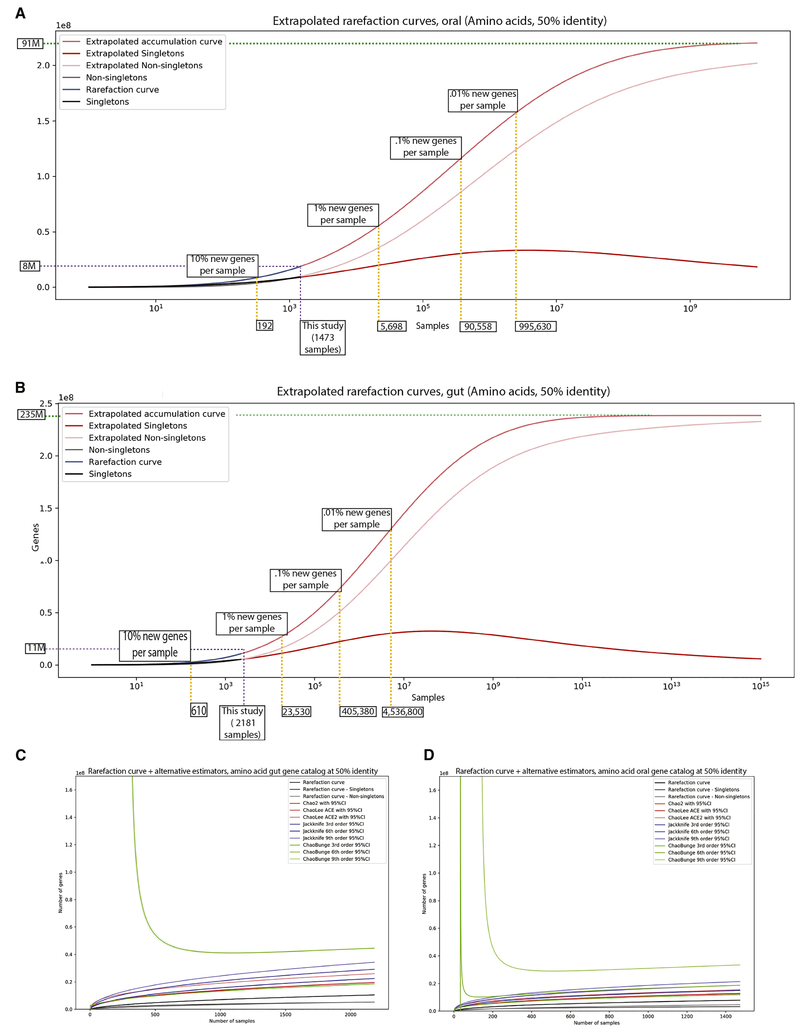
Despite substantial interest in the species diversity of the human microbiome and its role in disease, the scale of its genetic diversity, which is fundamental to deciphering human-microbe interactions, has not been quantified. Here, we conducted a cross-study meta-analysis of metagenomes from two human body niches, the mouth and gut, covering 3,655 samples from 13 studies. We found staggering genetic heterogeneity in the dataset, identifying a total of 45,666,334 non-redundant genes (23,961,508 oral and 22,254,436 gut) at the 95% identity level. Fifty percent of all genes were "singletons," or unique to a single metagenomic sample. Singletons were enriched for different functions (compared with non-singletons) and arose from sub-population-specific microbial strains. Overall, these results provide potential bases for the unexplained heterogeneity observed in microbiome-derived human phenotypes. One the basis of these data, we built a resource, which can be accessed at https://microbial-genes.bio.
Keywords: de novo assembly; gene catalog; gene diversity; gut microbiome; metagenomics; microbial diversity; oral microbiome.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
The authors have no competing interests to declare.
Figures
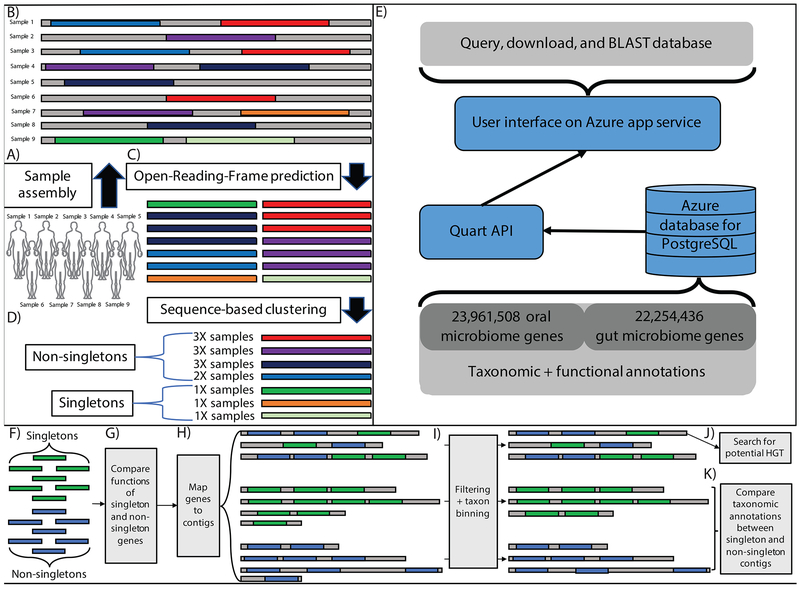
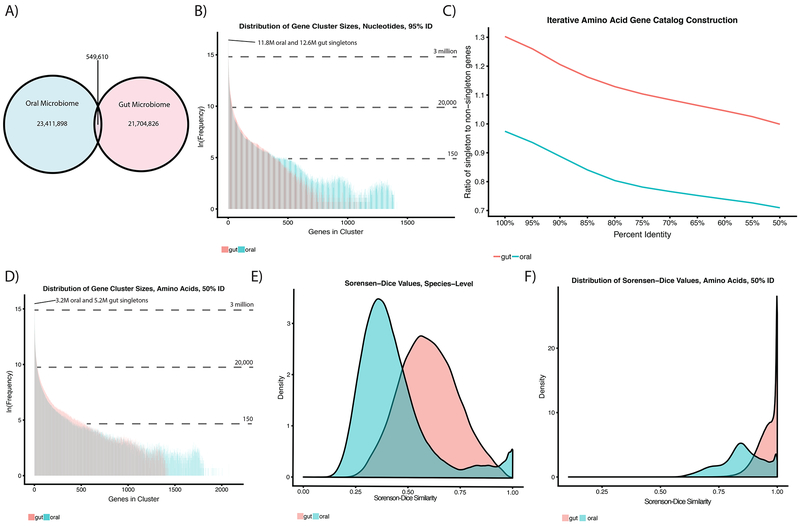
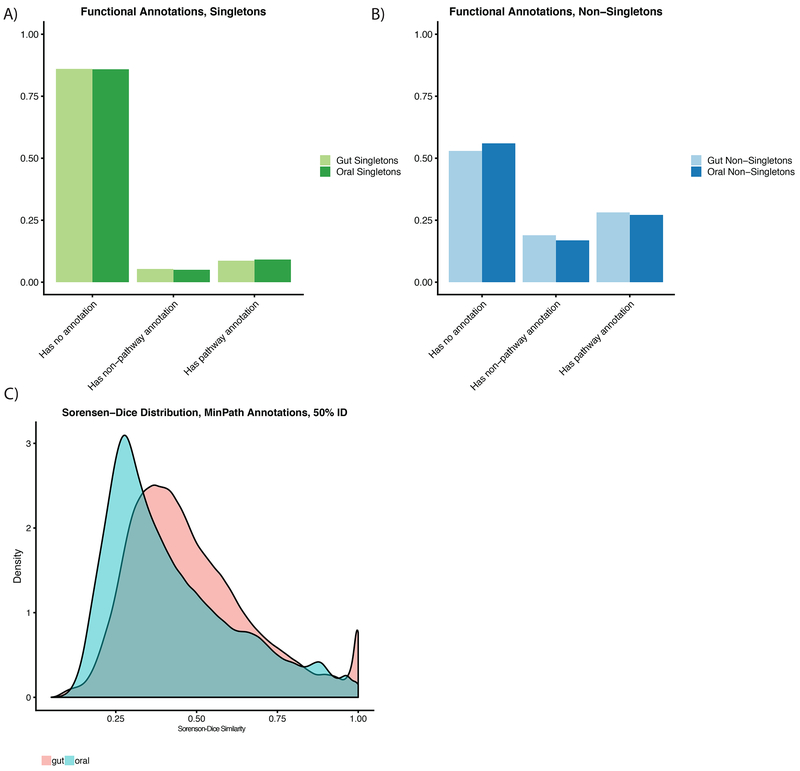
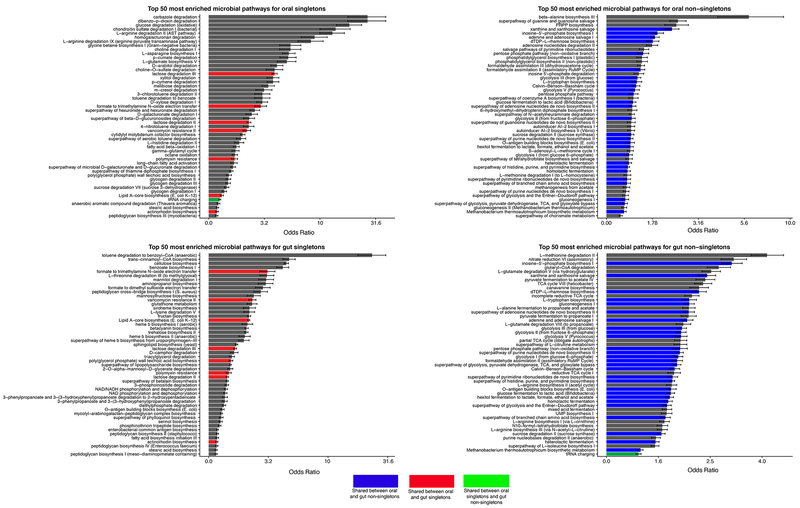
References
-
- Ayling Martin, Clark Matthew D., and Leggett Richard M.. 2018. “New Approaches for Assembly of Short-Read Metagenomic Data.” e27332v1 PeerJ Preprints. 10.7287/peerj.preprints.27332v1. - DOI
-
- Benjamini Yoav, and Yekutieli Daniel. 2001. “The Control of the False Discovery Rate in Multiple Testing under Dependency.” Annals of Statistics 29 (4): 1165–88.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
