

Mitochondria in the signaling pathways that control longevity and health span
- PMID: 31415807
- PMCID: PMC7479635
- DOI: 10.1016/j.arr.2019.100940
Mitochondria in the signaling pathways that control longevity and health span
Abstract
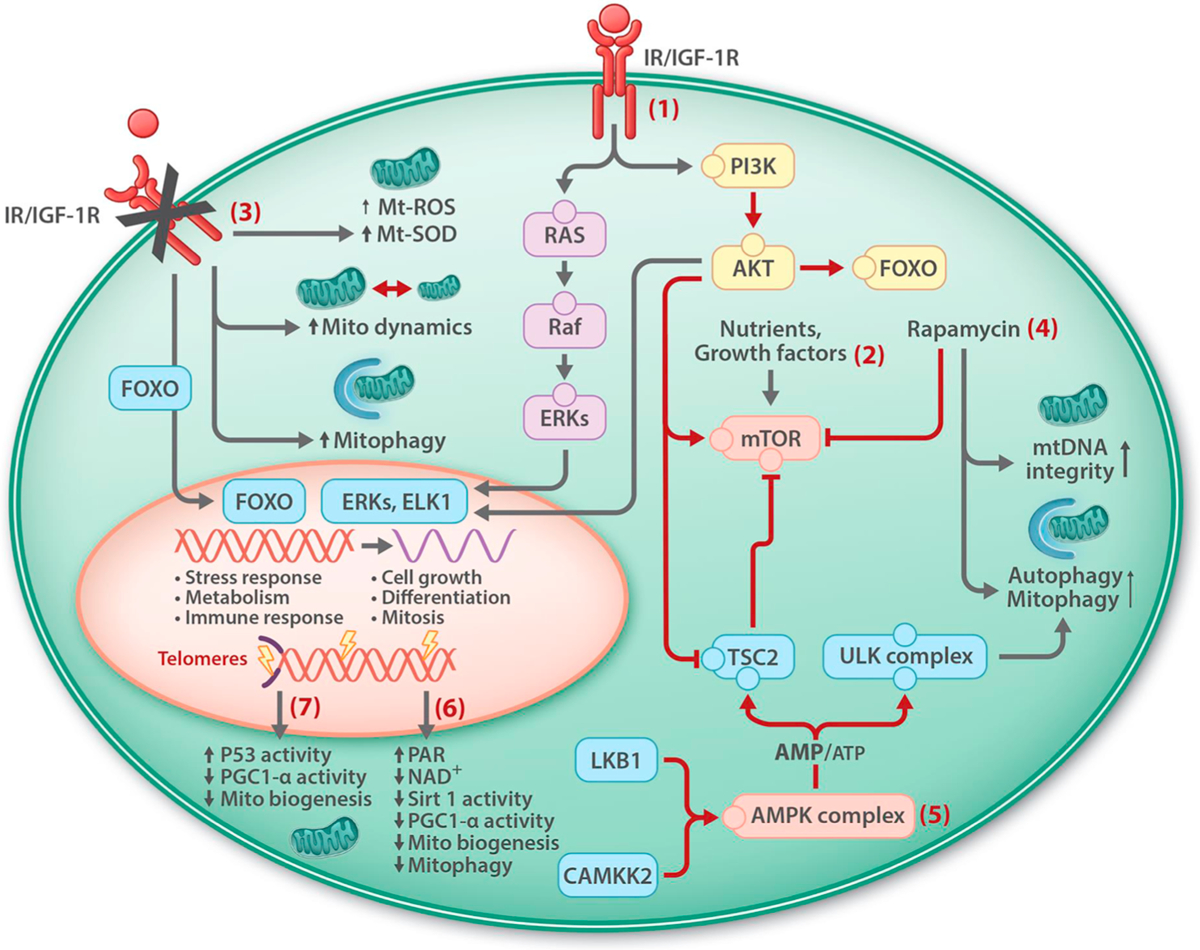
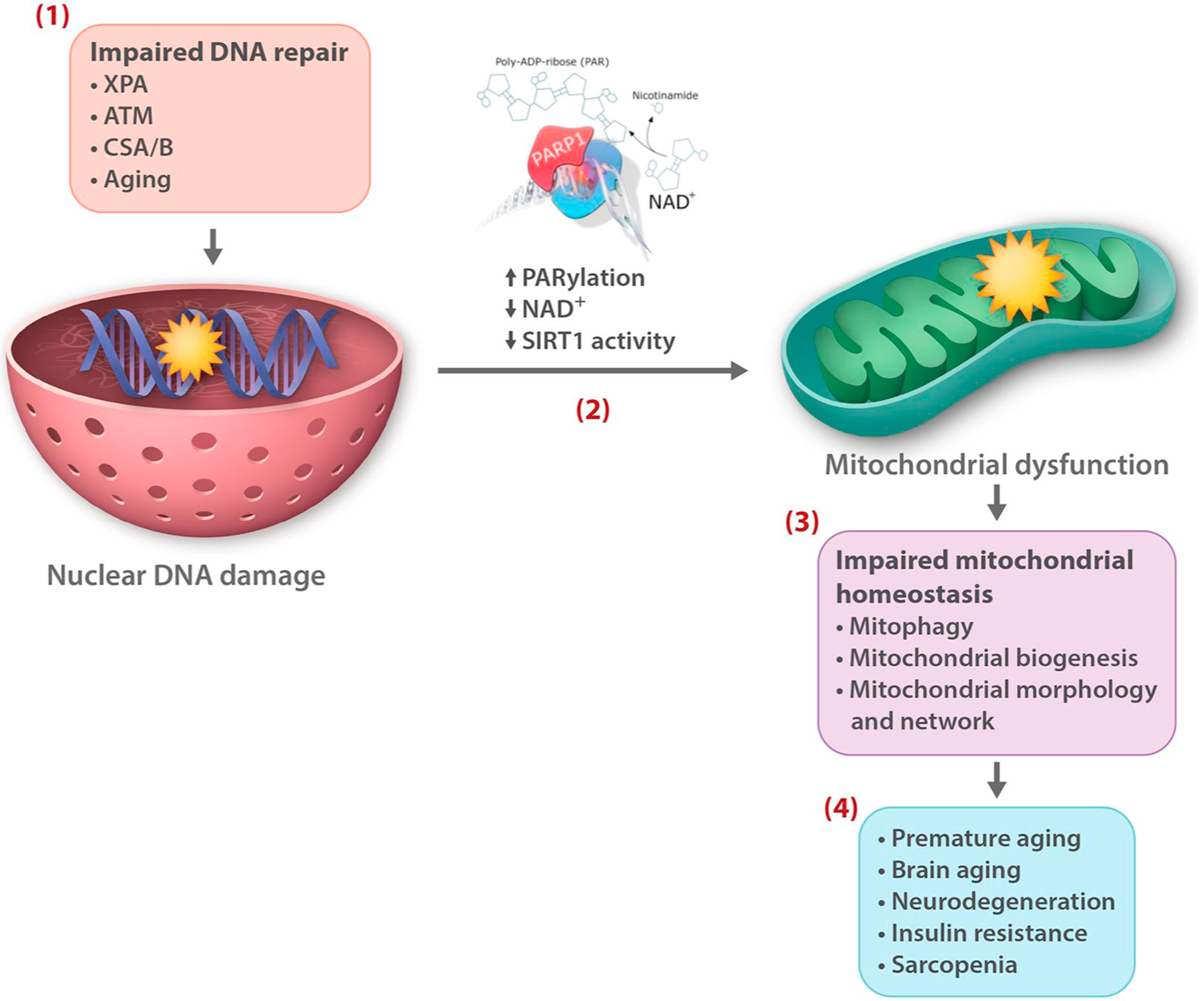
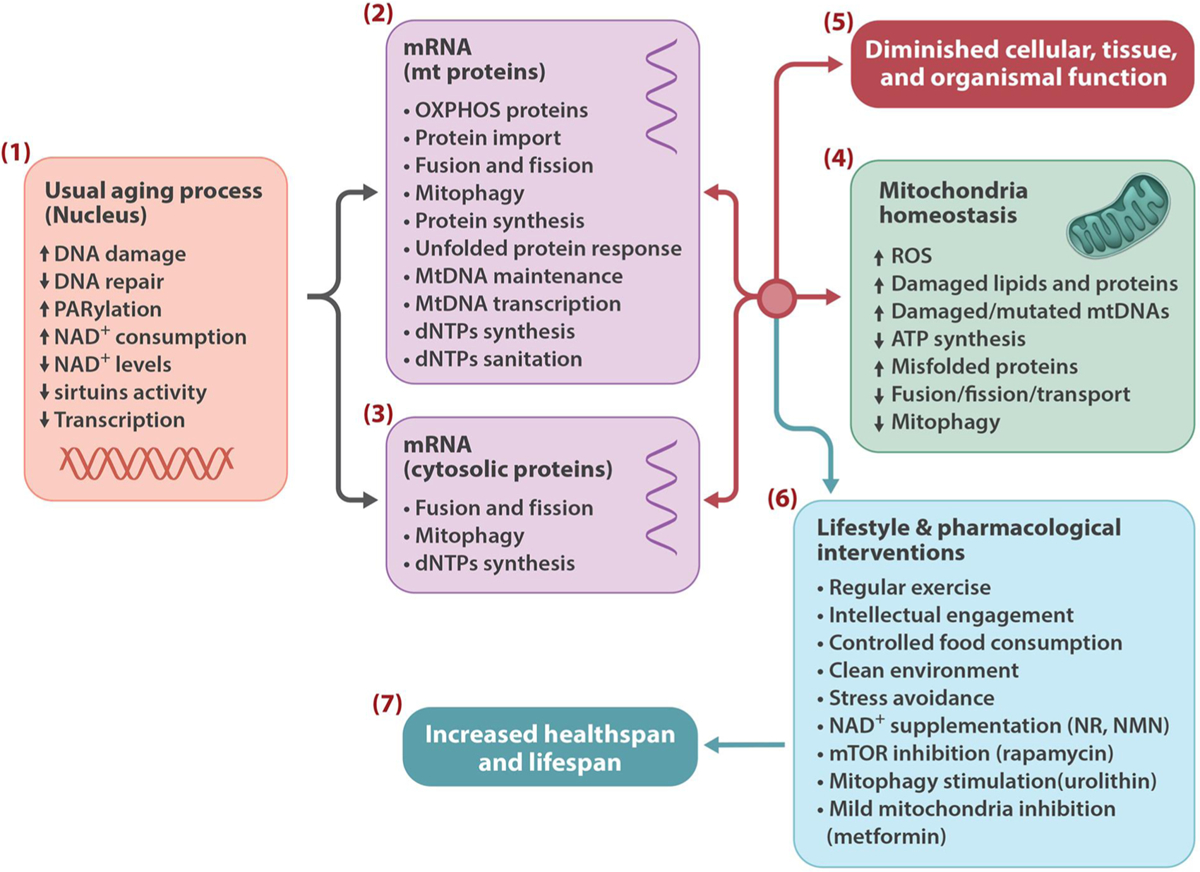
Genetic and pharmacological intervention studies have identified evolutionarily conserved and functionally interconnected networks of cellular energy homeostasis, nutrient-sensing, and genome damage response signaling pathways, as prominent regulators of longevity and health span in various species. Mitochondria are the primary sites of ATP production and are key players in several other important cellular processes. Mitochondrial dysfunction diminishes tissue and organ functional performance and is a commonly considered feature of the aging process. Here we review the evidence that through reciprocal and multilevel functional interactions, mitochondria are implicated in the lifespan modulation function of these pathways, which altogether constitute a highly dynamic and complex system that controls the aging process. An important characteristic of these pathways is their extensive crosstalk and apparent malleability to modification by non-invasive pharmacological, dietary, and lifestyle interventions, with promising effects on lifespan and health span in animal models and potentially also in humans.
Keywords: Aging; DNA repair; Energy; Lifespan; Metabolism; Mitochondria.
Published by Elsevier B.V.
Conflict of interest statement
Declaration of Competing Interest
None.
Figures
Similar articles
-
Mitochondrial stress signaling in longevity: a new role for mitochondrial function in aging.Redox Biol. 2014 Jul 27;2:936-44. doi: 10.1016/j.redox.2014.07.005. eCollection 2014. Redox Biol. 2014. PMID: 25180170 Free PMC article. Review.
-
The biochemistry and cell biology of aging: metabolic regulation through mitochondrial signaling.Am J Physiol Endocrinol Metab. 2014 Mar;306(6):E581-91. doi: 10.1152/ajpendo.00665.2013. Epub 2014 Jan 22. Am J Physiol Endocrinol Metab. 2014. PMID: 24452454 Review.
-
Prohibitin couples diapause signalling to mitochondrial metabolism during ageing in C. elegans.Nature. 2009 Oct 8;461(7265):793-7. doi: 10.1038/nature08466. Nature. 2009. PMID: 19812672
-
Energy, quiescence and the cellular basis of animal life spans.Comp Biochem Physiol A Mol Integr Physiol. 2006 Jan;143(1):12-23. doi: 10.1016/j.cbpa.2005.11.002. Epub 2005 Dec 27. Comp Biochem Physiol A Mol Integr Physiol. 2006. PMID: 16377223 Review.
-
Mitochondrial longevity pathways.Biochim Biophys Acta. 2011 Jan;1813(1):260-8. doi: 10.1016/j.bbamcr.2010.10.007. Epub 2010 Oct 13. Biochim Biophys Acta. 2011. PMID: 20950653 Review.
Cited by
-
Mitochondrial destabilization in tendinopathy and potential therapeutic strategies.J Orthop Translat. 2024 Oct 3;49:49-61. doi: 10.1016/j.jot.2024.09.003. eCollection 2024 Nov. J Orthop Translat. 2024. PMID: 39430132 Free PMC article. Review.
-
Hippocampal tau oligomerization early in tau pathology coincides with a transient alteration of mitochondrial homeostasis and DNA repair in a mouse model of tauopathy.Acta Neuropathol Commun. 2020 Mar 4;8(1):25. doi: 10.1186/s40478-020-00896-8. Acta Neuropathol Commun. 2020. PMID: 32131898 Free PMC article.
-
Aging conundrum: A perspective for ovarian aging.Front Endocrinol (Lausanne). 2022 Aug 19;13:952471. doi: 10.3389/fendo.2022.952471. eCollection 2022. Front Endocrinol (Lausanne). 2022. PMID: 36060963 Free PMC article. Review.
-
Cellular rejuvenation: molecular mechanisms and potential therapeutic interventions for diseases.Signal Transduct Target Ther. 2023 Mar 14;8(1):116. doi: 10.1038/s41392-023-01343-5. Signal Transduct Target Ther. 2023. PMID: 36918530 Free PMC article. Review.
-
Molecular mechanisms of coronary microvascular endothelial dysfunction in diabetes mellitus: focus on mitochondrial quality surveillance.Angiogenesis. 2022 Aug;25(3):307-329. doi: 10.1007/s10456-022-09835-8. Epub 2022 Mar 18. Angiogenesis. 2022. PMID: 35303170 Review.
References
-
- Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu M, Tian J, de Souza-Pinto NC, Scheibye-Knudsen M, Wilson DM 3rd, Stevnsner T, Bohr VA, 2010. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 24, 2334–2346. - PMC - PubMed
-
- Ahmed S, Passos JF, Birket MJ, Beckmann T, Brings S, Peters H, Birch-Machin MA, von Zglinicki T, Saretzki G, 2008. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J. Cell. Sci 121, 1046–1053. - PubMed
-
- Akbari M, Krokan HE, 2008. Cytotoxicity and mutagenicity of endogenous DNA base lesions as potential cause of human aging. Mech. Ageing Dev 129, 353–365. - PubMed
-
- Akbari M, Pena-Diaz J, Andersen S, Liabakk NB, Otterlei M, Krokan HE, 2009. Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair (Amst.) 8, 834–843. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
