

Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: just What It Takes to Make a Blood Vessel
- PMID: 31416282
- PMCID: PMC6721072
- DOI: 10.3390/ijms20163962
Endothelial Ca2+ Signaling, Angiogenesis and Vasculogenesis: just What It Takes to Make a Blood Vessel
Abstract
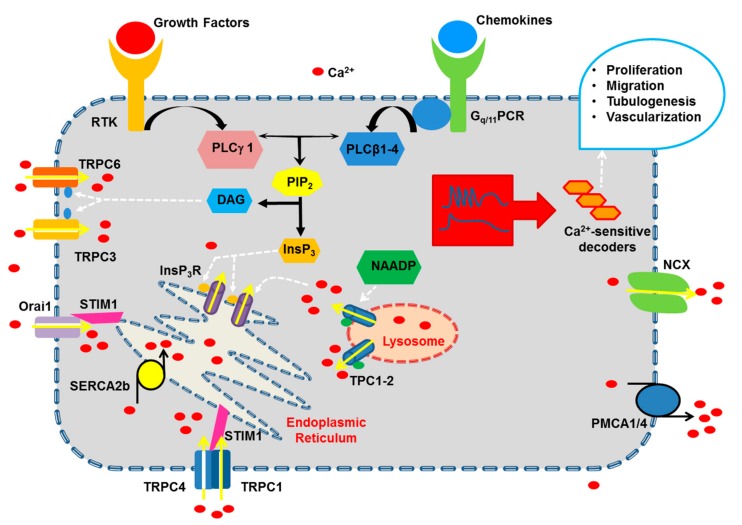
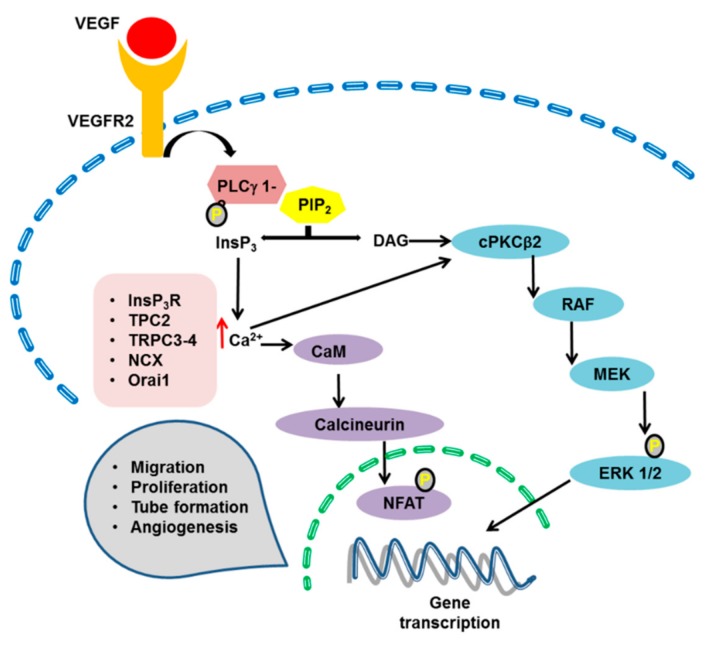
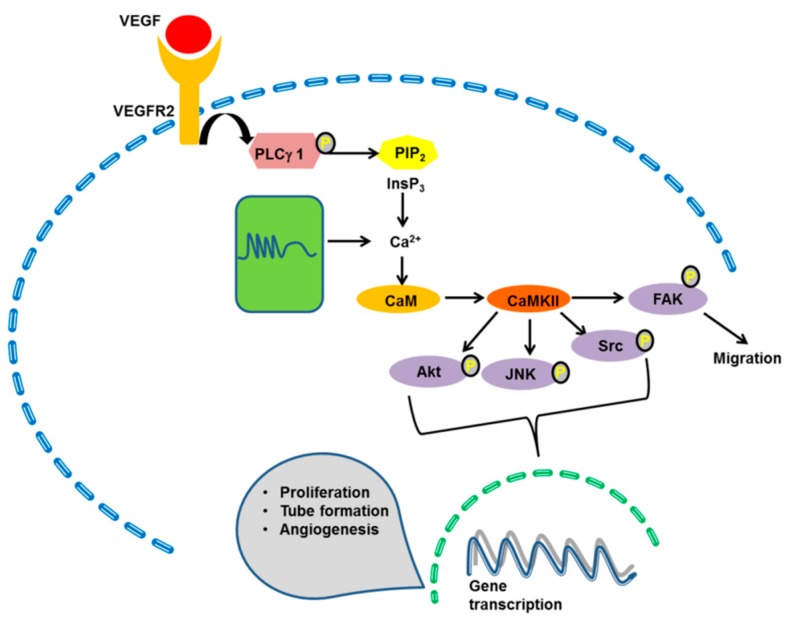
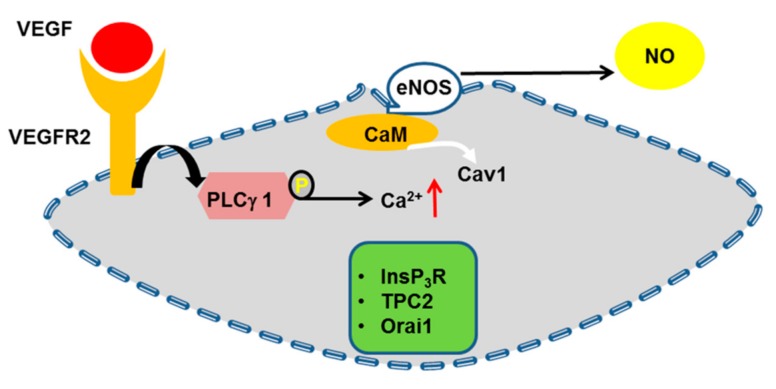
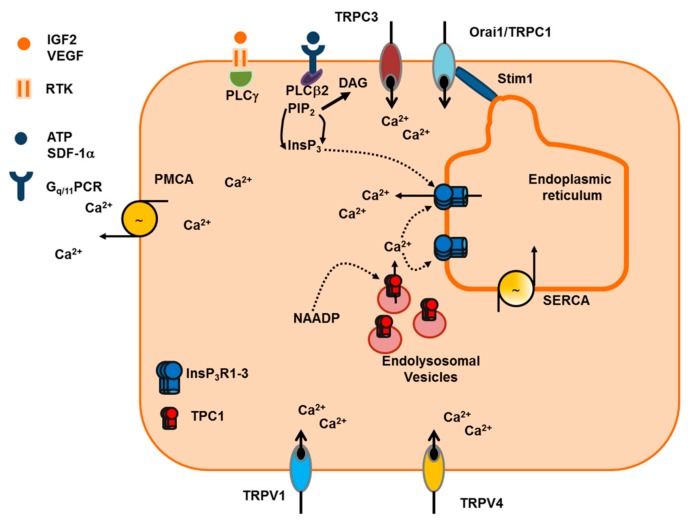
It has long been known that endothelial Ca2+ signals drive angiogenesis by recruiting multiple Ca2+-sensitive decoders in response to pro-angiogenic cues, such as vascular endothelial growth factor, basic fibroblast growth factor, stromal derived factor-1α and angiopoietins. Recently, it was shown that intracellular Ca2+ signaling also drives vasculogenesis by stimulation proliferation, tube formation and neovessel formation in endothelial progenitor cells. Herein, we survey how growth factors, chemokines and angiogenic modulators use endothelial Ca2+ signaling to regulate angiogenesis and vasculogenesis. The endothelial Ca2+ response to pro-angiogenic cues may adopt different waveforms, ranging from Ca2+ transients or biphasic Ca2+ signals to repetitive Ca2+ oscillations, and is mainly driven by endogenous Ca2+ release through inositol-1,4,5-trisphosphate receptors and by store-operated Ca2+ entry through Orai1 channels. Lysosomal Ca2+ release through nicotinic acid adenine dinucleotide phosphate-gated two-pore channels is, however, emerging as a crucial pro-angiogenic pathway, which sustains intracellular Ca2+ mobilization. Understanding how endothelial Ca2+ signaling regulates angiogenesis and vasculogenesis could shed light on alternative strategies to induce therapeutic angiogenesis or interfere with the aberrant vascularization featuring cancer and intraocular disorders.
Keywords: TRPC channels.; basic fibroblast growth factor; endothelial cells; endothelial colony forming cells; inositol-1,4,5-trisphosphate; nicotinic acid adenine dinucleotide phosphate; store-operated Ca2+ entry; stromal derived factor-1α; vascular endothelial growth factor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
