

Functional Transcriptome Analysis in ARSACS KO Cell Model Reveals a Role of Sacsin in Autophagy
- PMID: 31417125
- PMCID: PMC6695435
- DOI: 10.1038/s41598-019-48047-x
Functional Transcriptome Analysis in ARSACS KO Cell Model Reveals a Role of Sacsin in Autophagy
Abstract
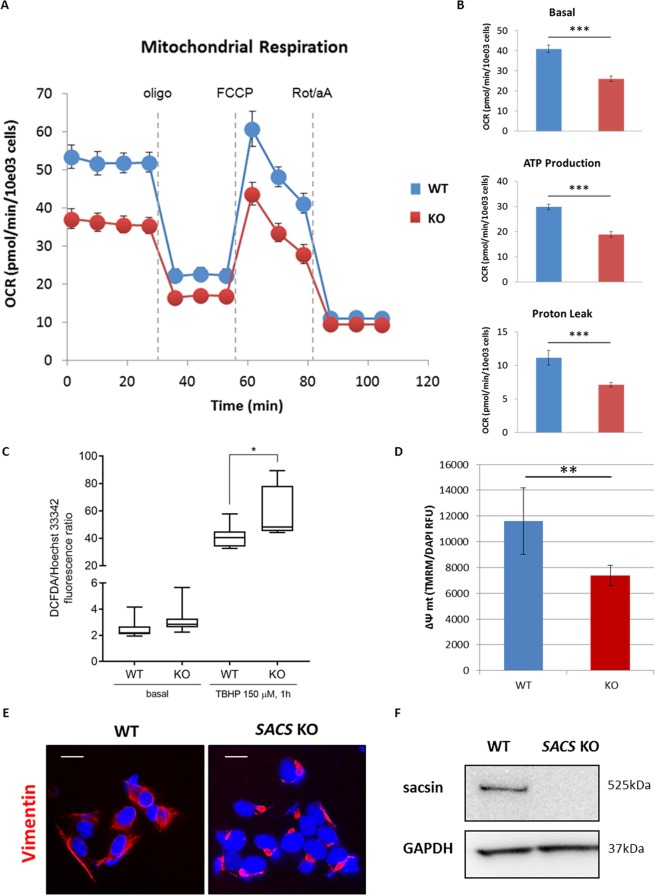
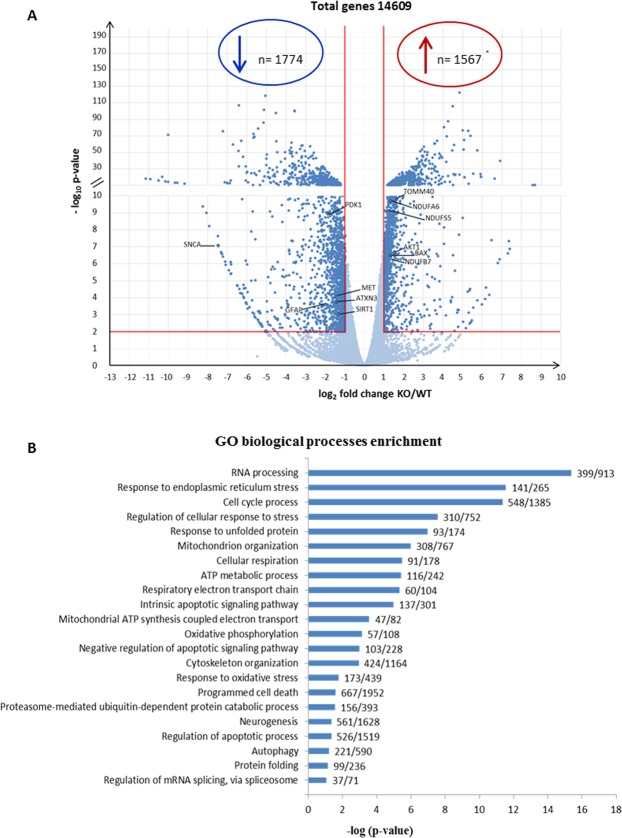
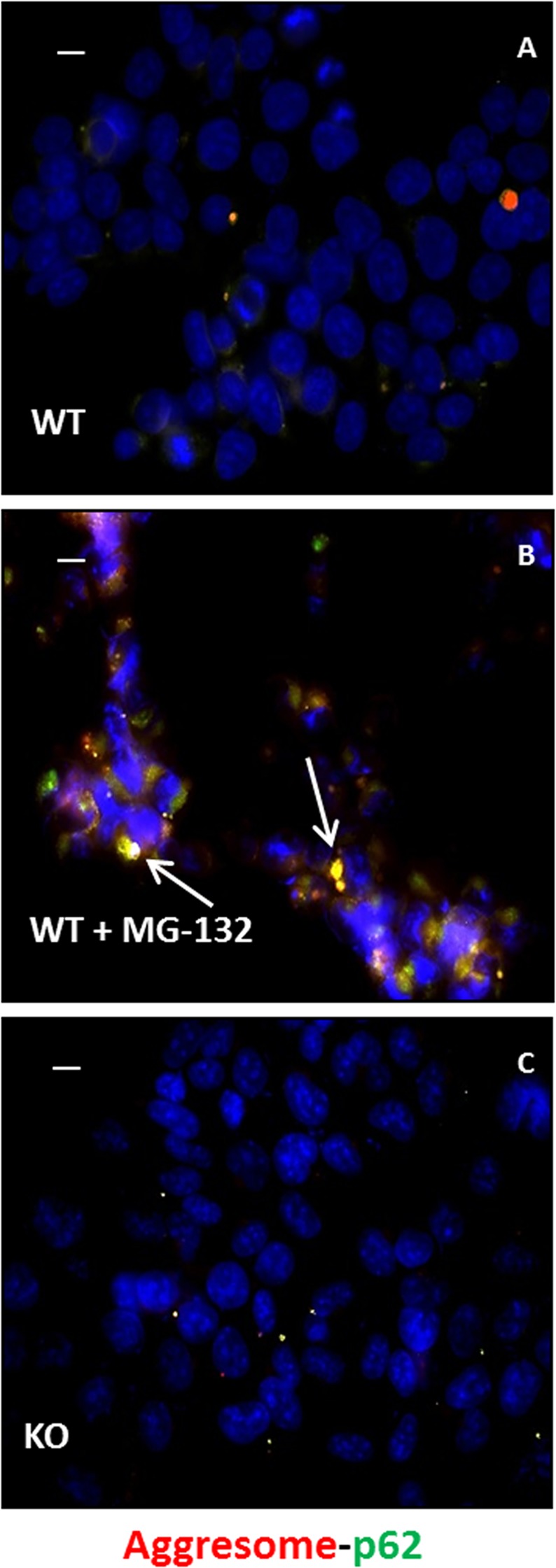
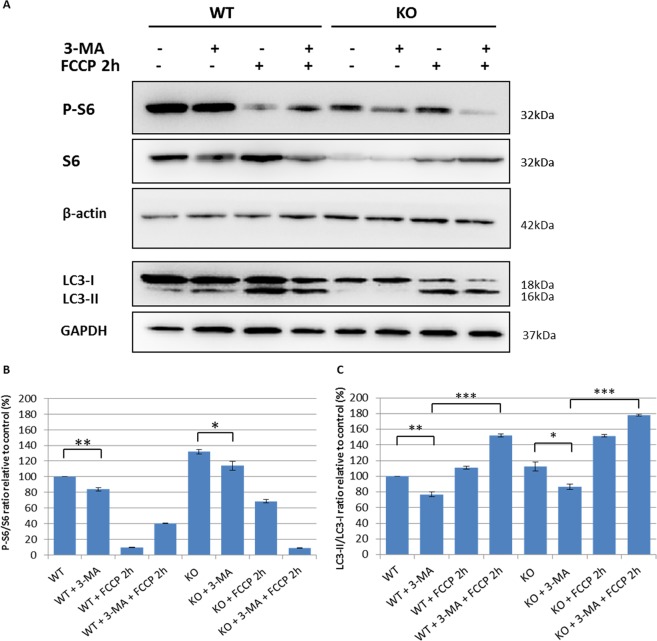
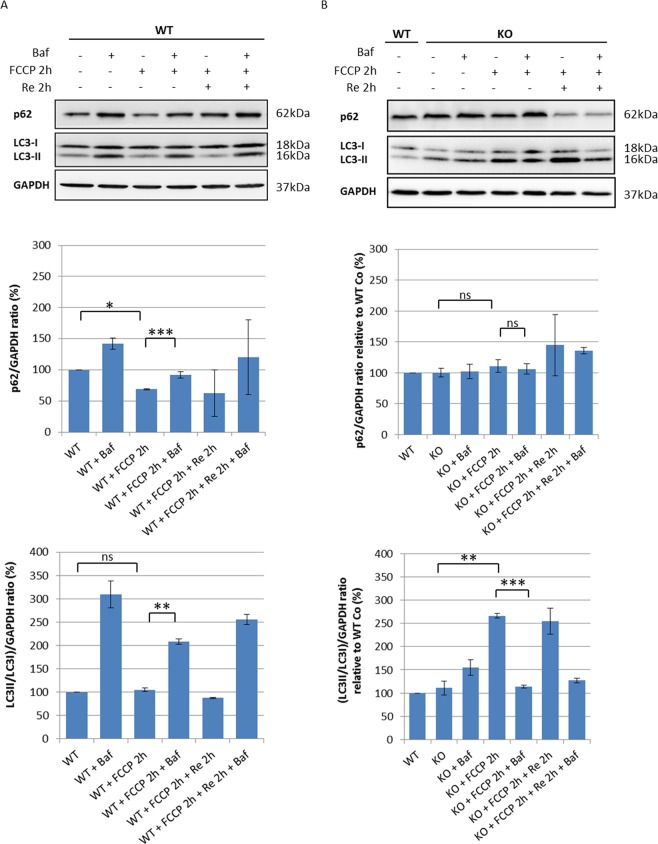
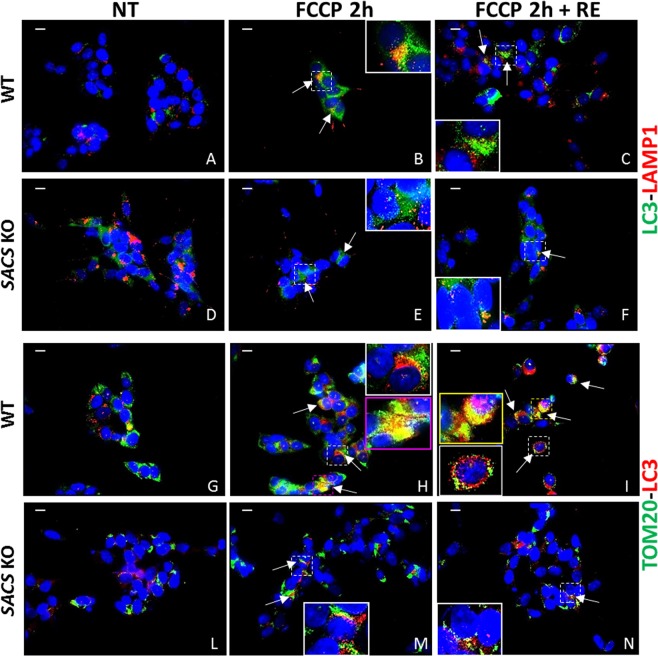
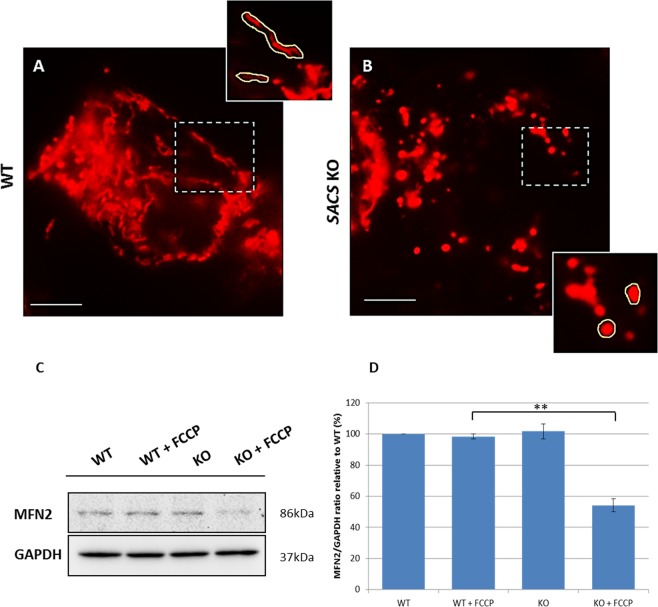
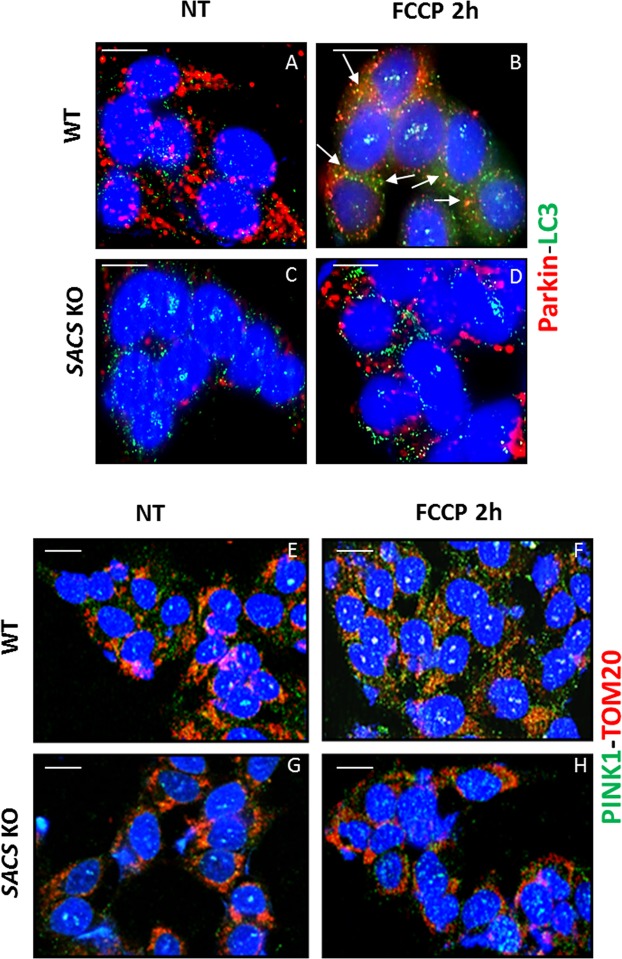
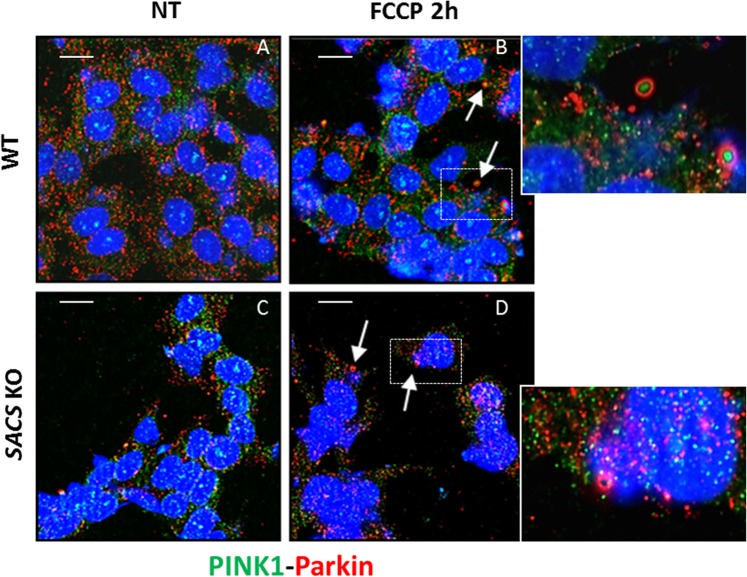
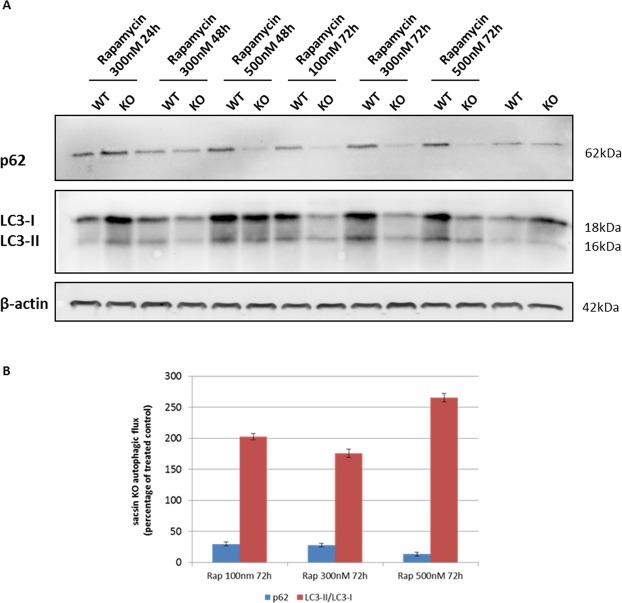
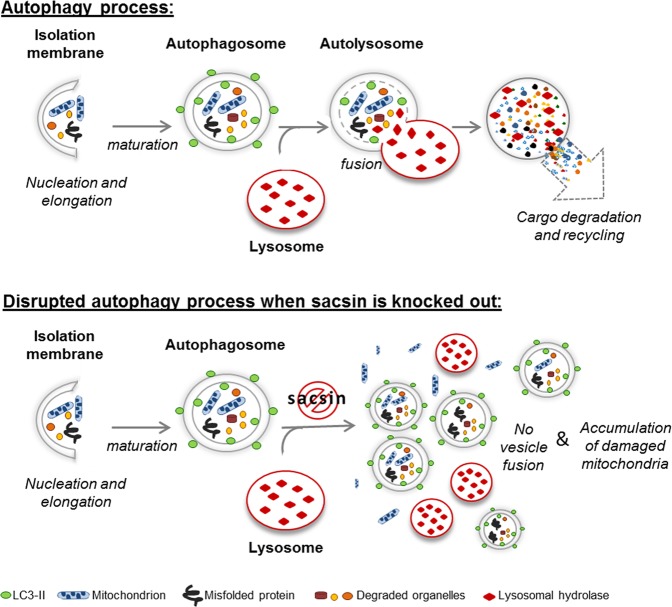
Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) is a rare early-onset neurological disease caused by mutations in SACS, which encodes sacsin. The complex architecture of sacsin suggests that it could be a key player in cellular protein quality control system. Molecular chaperones that operate in protein folding/unfolding and assembly/disassembly patterns have been described as essential modulators of selectivity during the autophagy process. We performed RNA-sequencing analysis to generate a whole-genome molecular signature profile of sacsin knockout cells. Using data analysis of biological processes significantly disrupted due to loss of sacsin, we confirmed the presence of decreased mitochondrial function associated with increased oxidative stress, and also provided a demonstration of a defective autophagic pathway in sacsin-depleted cells. Western blotting assays revealed decreased expression of LC3 and increased levels of p62 even after treatment with the lysosomal inhibitor bafilomycin A1, indicating impairment of the autophagic flux. Moreover, we found reduced co-immunolocalization of the autophagosome marker LC3 with lysosomal and mitochondrial markers suggesting fusion inhibition of autophagic compartments and subsequent failed cargo degradation, in particular failed degradation of damaged mitochondria. Pharmacological up-regulation of autophagy restored correct autophagic flux in sacsin knockout cells. These results corroborate the hypothesis that sacsin may play a role in autophagy. Chemical manipulation of this pathway might represent a new target to alleviate clinical and pathological symptoms, delaying the processes of neurodegeneration in ARSACS.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
