

Guidelines for evaluating myocardial cell death
- PMID: 31418596
- PMCID: PMC6879915
- DOI: 10.1152/ajpheart.00259.2019
Guidelines for evaluating myocardial cell death
Erratum in
-
Corrigendum.Am J Physiol Heart Circ Physiol. 2019 Dec 1;317(6):H1390. doi: 10.1152/ajpheart.zh4-2975-corr.2019. Am J Physiol Heart Circ Physiol. 2019. PMID: 31797698 Free PMC article. No abstract available.
Abstract
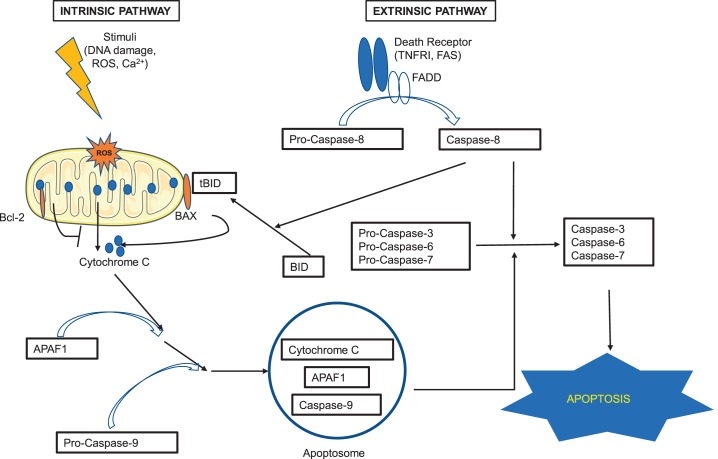
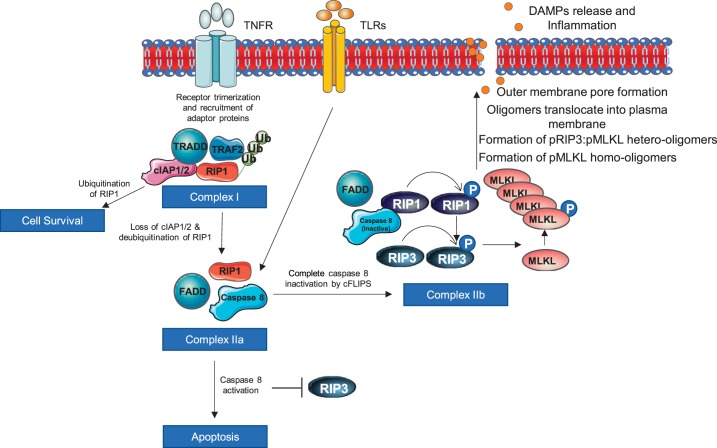
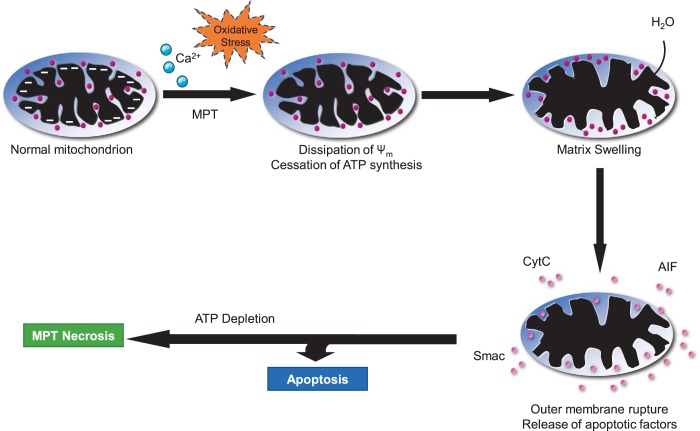
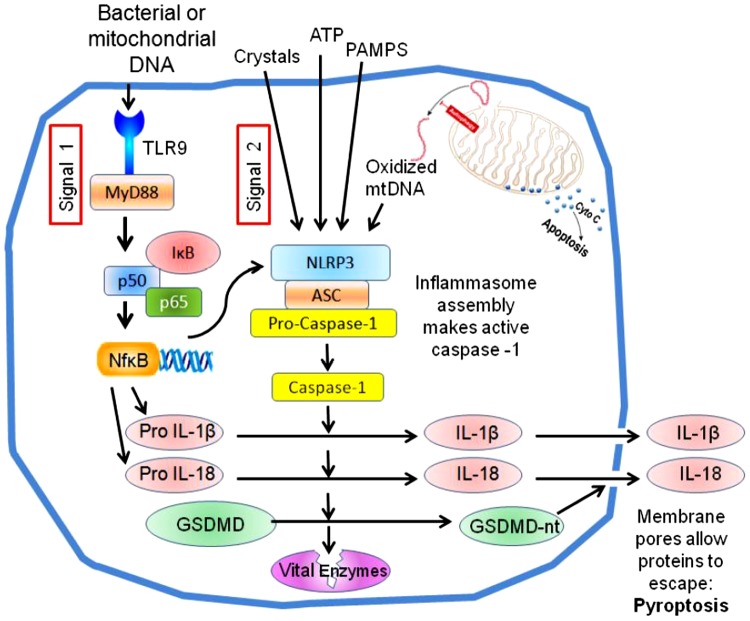
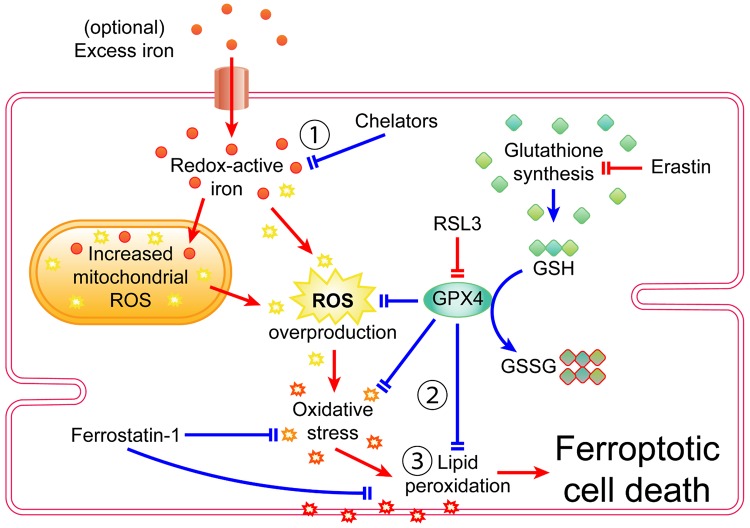
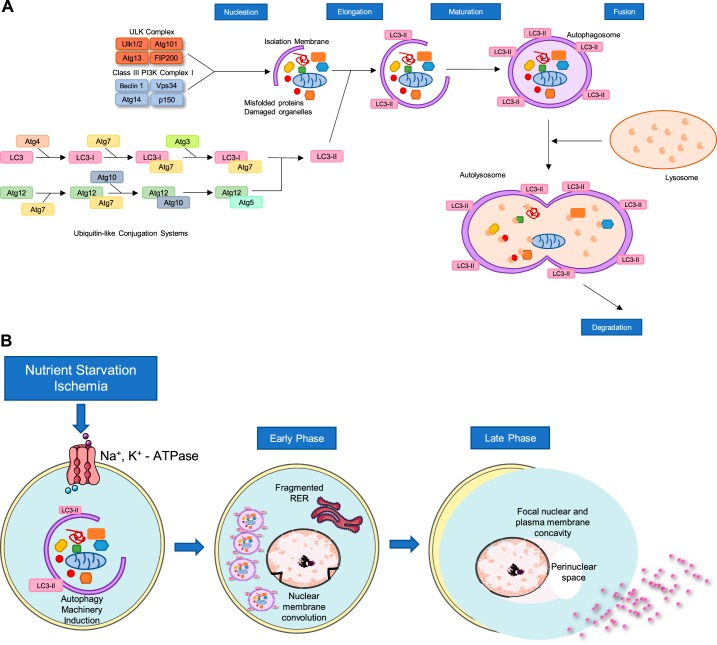
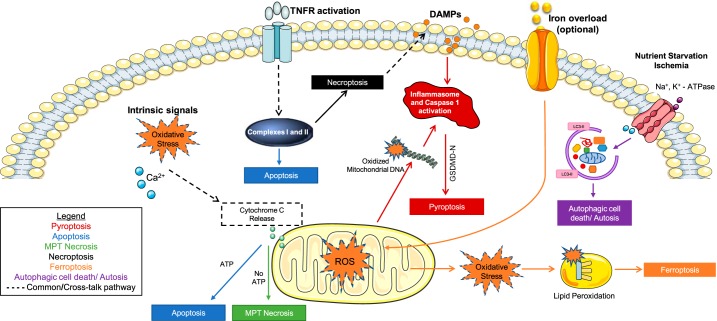
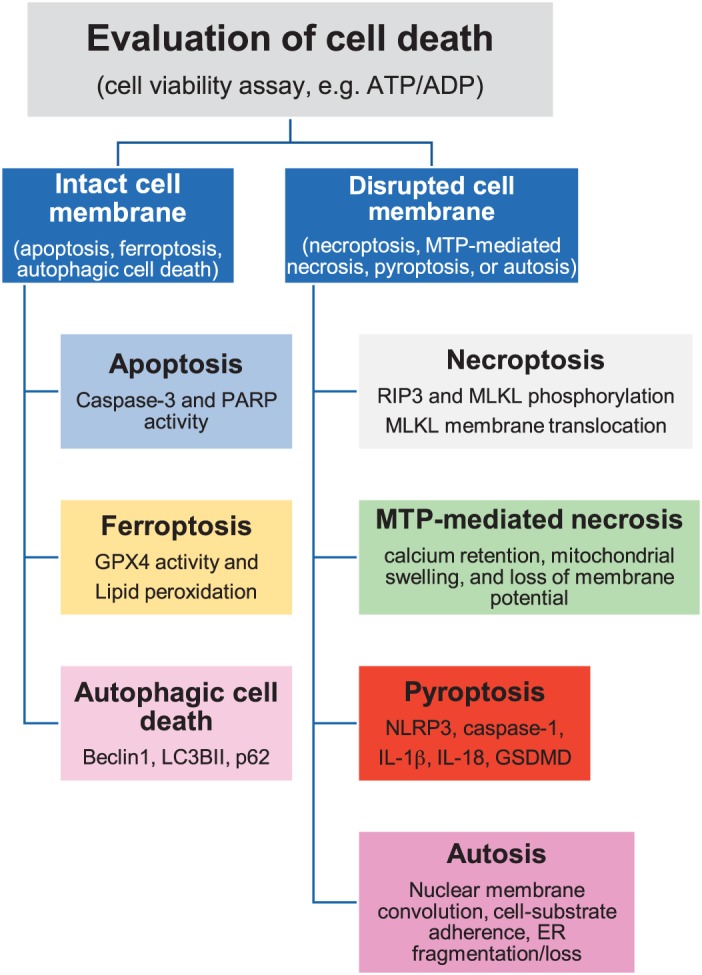
Cell death is a fundamental process in cardiac pathologies. Recent studies have revealed multiple forms of cell death, and several of them have been demonstrated to underlie adverse cardiac remodeling and heart failure. With the expansion in the area of myocardial cell death and increasing concerns over rigor and reproducibility, it is important and timely to set a guideline for the best practices of evaluating myocardial cell death. There are six major forms of regulated cell death observed in cardiac pathologies, namely apoptosis, necroptosis, mitochondrial-mediated necrosis, pyroptosis, ferroptosis, and autophagic cell death. In this article, we describe the best methods to identify, measure, and evaluate these modes of myocardial cell death. In addition, we discuss the limitations of currently practiced myocardial cell death mechanisms.
Keywords: apoptosis; autophagic cell death; cardiovascular disease; ferroptosis; heart; mitochondrial-mediated necrosis; necroptosis; pyroptosis.
Conflict of interest statement
A. Abbate has served as a consultant to Astra-Zeneca, Janssen, Merck, Novartis, Olatec and Serpin pharma.
Figures
References
-
- Abbate A, De Falco M, Morales C, Gelpi RJ, Prisco M, De Luca A, Palleiro J, Fedele V, Feroce F, Baldi F, Vetrovec GW, Baldi A. Electron microscopy characterization of cardiomyocyte apoptosis in ischemic heart disease. Int J Cardiol 114: 118–120, 2007. doi: 10.1016/j.ijcard.2005.11.025. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources