

Bi-allelic GOT2 Mutations Cause a Treatable Malate-Aspartate Shuttle-Related Encephalopathy
- PMID: 31422819
- PMCID: PMC6732527
- DOI: 10.1016/j.ajhg.2019.07.015
Bi-allelic GOT2 Mutations Cause a Treatable Malate-Aspartate Shuttle-Related Encephalopathy
Abstract
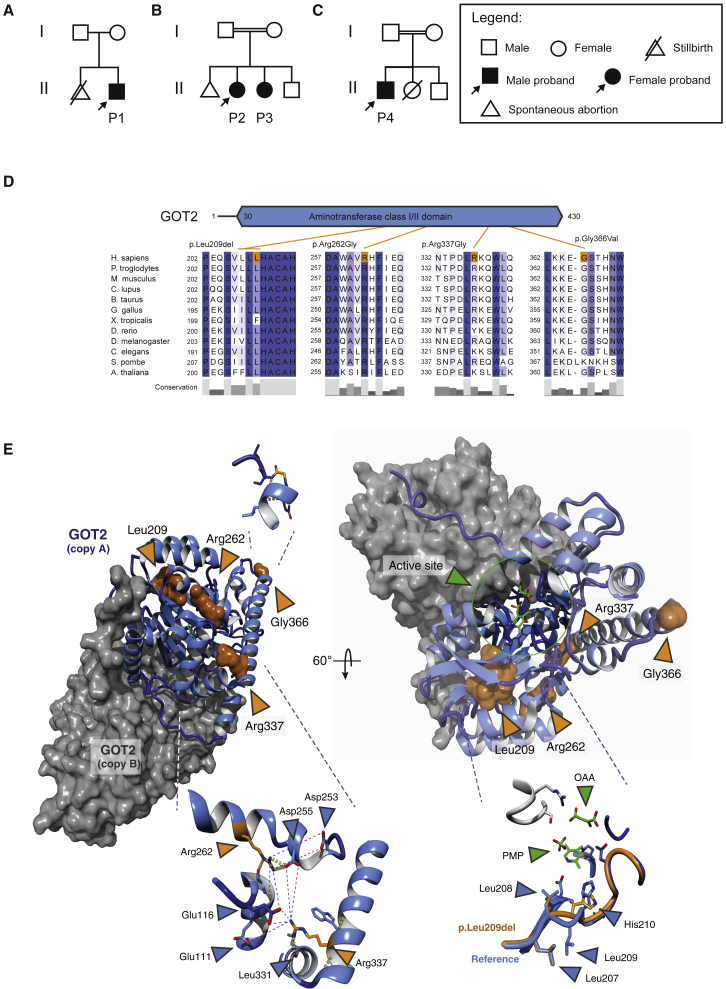
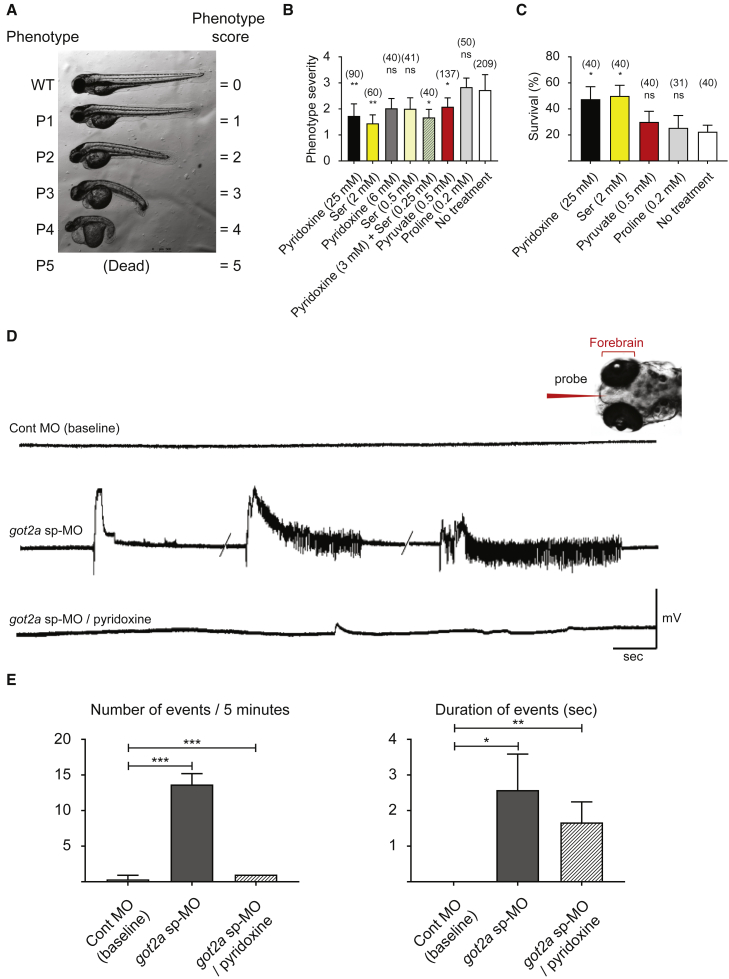
Early-infantile encephalopathies with epilepsy are devastating conditions mandating an accurate diagnosis to guide proper management. Whole-exome sequencing was used to investigate the disease etiology in four children from independent families with intellectual disability and epilepsy, revealing bi-allelic GOT2 mutations. In-depth metabolic studies in individual 1 showed low plasma serine, hypercitrullinemia, hyperlactatemia, and hyperammonemia. The epilepsy was serine and pyridoxine responsive. Functional consequences of observed mutations were tested by measuring enzyme activity and by cell and animal models. Zebrafish and mouse models were used to validate brain developmental and functional defects and to test therapeutic strategies. GOT2 encodes the mitochondrial glutamate oxaloacetate transaminase. GOT2 enzyme activity was deficient in fibroblasts with bi-allelic mutations. GOT2, a member of the malate-aspartate shuttle, plays an essential role in the intracellular NAD(H) redox balance. De novo serine biosynthesis was impaired in fibroblasts with GOT2 mutations and GOT2-knockout HEK293 cells. Correcting the highly oxidized cytosolic NAD-redox state by pyruvate supplementation restored serine biosynthesis in GOT2-deficient cells. Knockdown of got2a in zebrafish resulted in a brain developmental defect associated with seizure-like electroencephalography spikes, which could be rescued by supplying pyridoxine in embryo water. Both pyridoxine and serine synergistically rescued embryonic developmental defects in zebrafish got2a morphants. The two treated individuals reacted favorably to their treatment. Our data provide a mechanistic basis for the biochemical abnormalities in GOT2 deficiency that may also hold for other MAS defects.
Keywords: EC 2.6.1.1.; GOT2; aspartate aminotransferase; encephalopathy; inborn error of metabolism; malate-aspartate shuttle; mitochondriopathy; pyridoxine responsive epilepsy; redox imbalance; treatment.
Crown Copyright © 2019. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Dawson A.G. Oxidation of cytosolic NADH formed during aerobic metabolism in mammalian cells. Trends Biochem. Sci. 1979;4:171–176.
-
- Purvis J.L., Lowenstein J.M. The relation between intra- and extramitochondrial pyridine nucleotides. J. Biol. Chem. 1961;236:2794–2803. - PubMed
-
- Wibom R., Lasorsa F.M., Töhönen V., Barbaro M., Sterky F.H., Kucinski T., Naess K., Jonsson M., Pierri C.L., Palmieri F., Wedell A. AGC1 deficiency associated with global cerebral hypomyelination. N. Engl. J. Med. 2009;361:489–495. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
