

De novo profiling of RNA viruses in Anopheles malaria vector mosquitoes from forest ecological zones in Senegal and Cambodia
- PMID: 31429704
- PMCID: PMC6702732
- DOI: 10.1186/s12864-019-6034-1
De novo profiling of RNA viruses in Anopheles malaria vector mosquitoes from forest ecological zones in Senegal and Cambodia
Erratum in
-
Correction to: De novo profiling of RNA viruses in Anopheles malaria vector mosquitoes from forest ecological zones in Senegal and Cambodia.BMC Genomics. 2019 Sep 5;20(1):698. doi: 10.1186/s12864-019-6067-5. BMC Genomics. 2019. PMID: 31488060 Free PMC article.
Abstract
Background: Mosquitoes are colonized by a large but mostly uncharacterized natural virome of RNA viruses, and the composition and distribution of the natural RNA virome may influence the biology and immunity of Anopheles malaria vector populations.
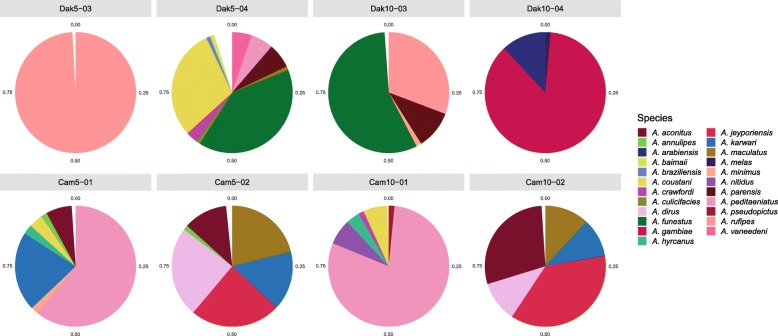
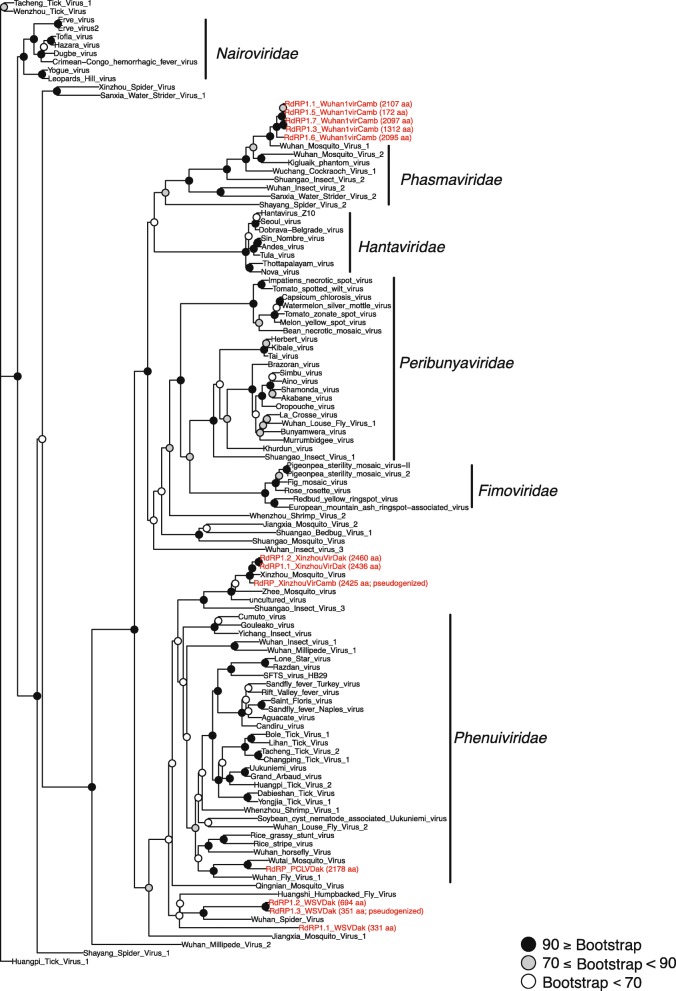
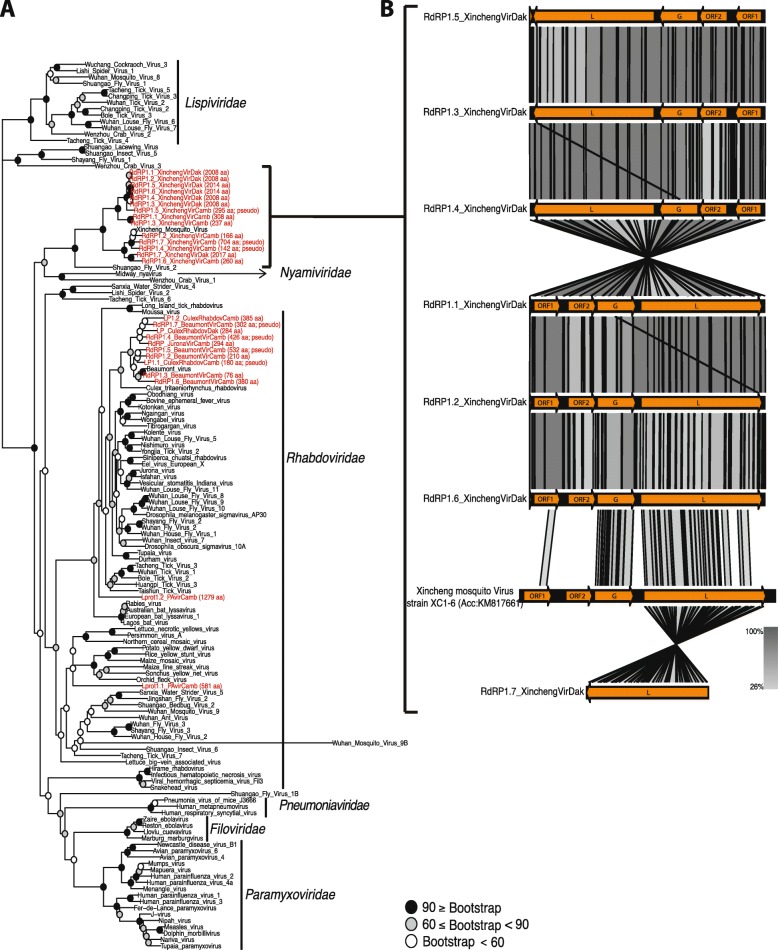
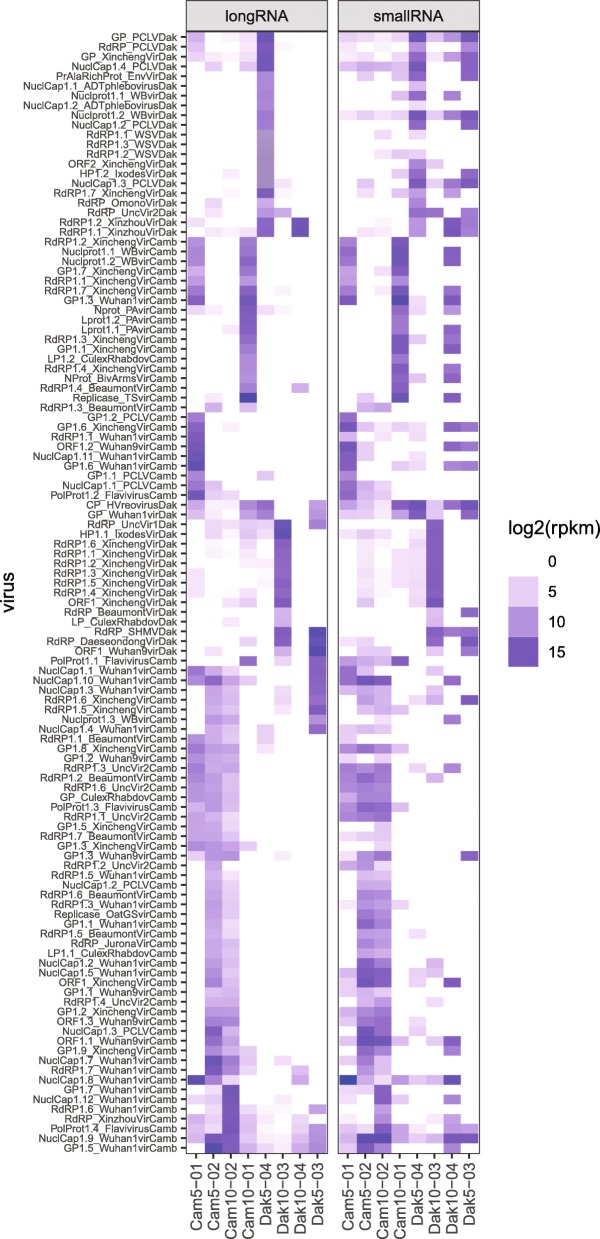
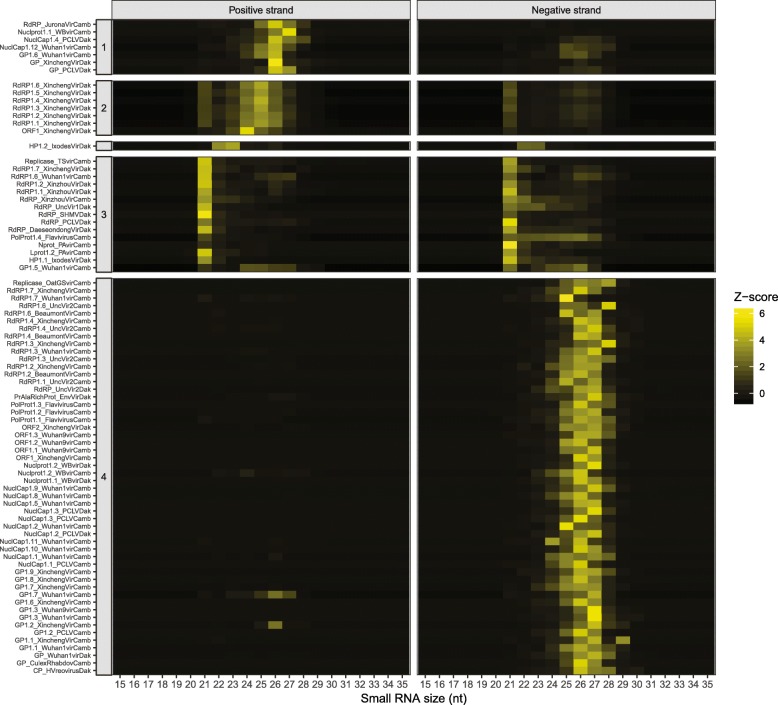
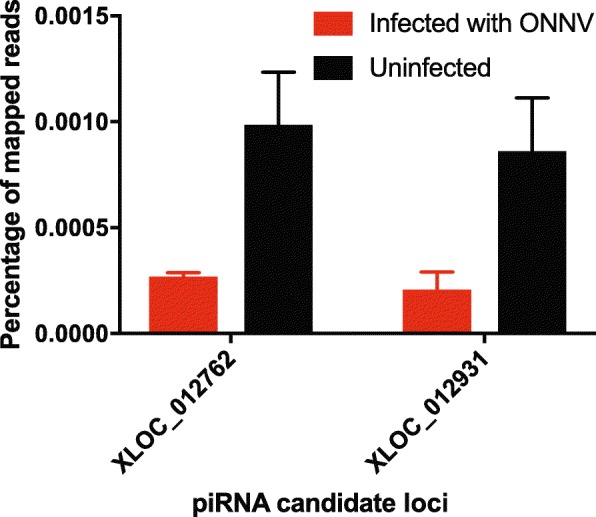
Results: Anopheles mosquitoes were sampled in malaria endemic forest village sites in Senegal and Cambodia, including Anopheles funestus, Anopheles gambiae group sp., and Anopheles coustani in Senegal, and Anopheles hyrcanus group sp., Anopheles maculatus group sp., and Anopheles dirus in Cambodia. The most frequent mosquito species sampled at both study sites are human malaria vectors. Small and long RNA sequences were depleted of mosquito host sequences, de novo assembled and clustered to yield non-redundant contigs longer than 500 nucleotides. Analysis of the assemblies by sequence similarity to known virus families yielded 115 novel virus sequences, and evidence supports a functional status for at least 86 of the novel viral contigs. Important monophyletic virus clades in the Bunyavirales and Mononegavirales orders were found in these Anopheles from Africa and Asia. The remaining non-host RNA assemblies that were unclassified by sequence similarity to known viruses were clustered by small RNA profiles, and 39 high-quality independent contigs strongly matched a pattern of classic RNAi processing of viral replication intermediates, suggesting they are entirely undescribed viruses. One thousand five hundred sixty-six additional high-quality unclassified contigs matched a pattern consistent with Piwi-interacting RNAs (piRNAs), suggesting that strand-biased piRNAs are generated from the natural virome in Anopheles. To functionally query piRNA effect, we analyzed piRNA expression in Anopheles coluzzii after infection with O'nyong nyong virus (family Togaviridae), and identified two piRNAs that appear to display specifically altered abundance upon arbovirus infection.
Conclusions: Anopheles vectors of human malaria in Africa and Asia are ubiquitously colonized by RNA viruses, some of which are monophyletic but clearly diverged from other arthropod viruses. The interplay between small RNA pathways, immunity, and the virome may represent part of the homeostatic mechanism maintaining virome members in a commensal or nonpathogenic state, and could potentially influence vector competence.
Keywords: Anopheles; Insect specific virus; Malaria vector; RNA virus; Virome; Virus genome assembly.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- World Health Organization . World Malaria Report 2017. Geneva: World Health Organization; 2017.
-
- Colmant AMG, Hobson-Peters J, Bielefeldt-Ohmann H, van den Hurk AF, Hall-Mendelin S, Chow WK, Johansen CA, Fros J, Simmonds P, Watterson D, et al. A new clade of insect-specific flaviviruses from Australian anopheles mosquitoes displays species-specific host restriction. mSphere. 2017;2(4):1–19. - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
