

Salt-inducible kinases dictate parathyroid hormone 1 receptor action in bone development and remodeling
- PMID: 31430259
- PMCID: PMC6877304
- DOI: 10.1172/JCI130126
Salt-inducible kinases dictate parathyroid hormone 1 receptor action in bone development and remodeling
Abstract
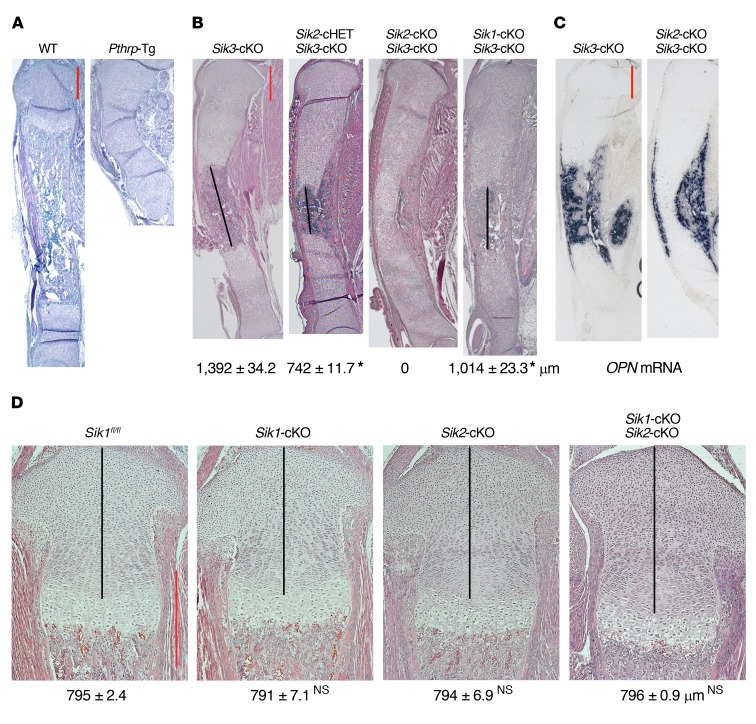
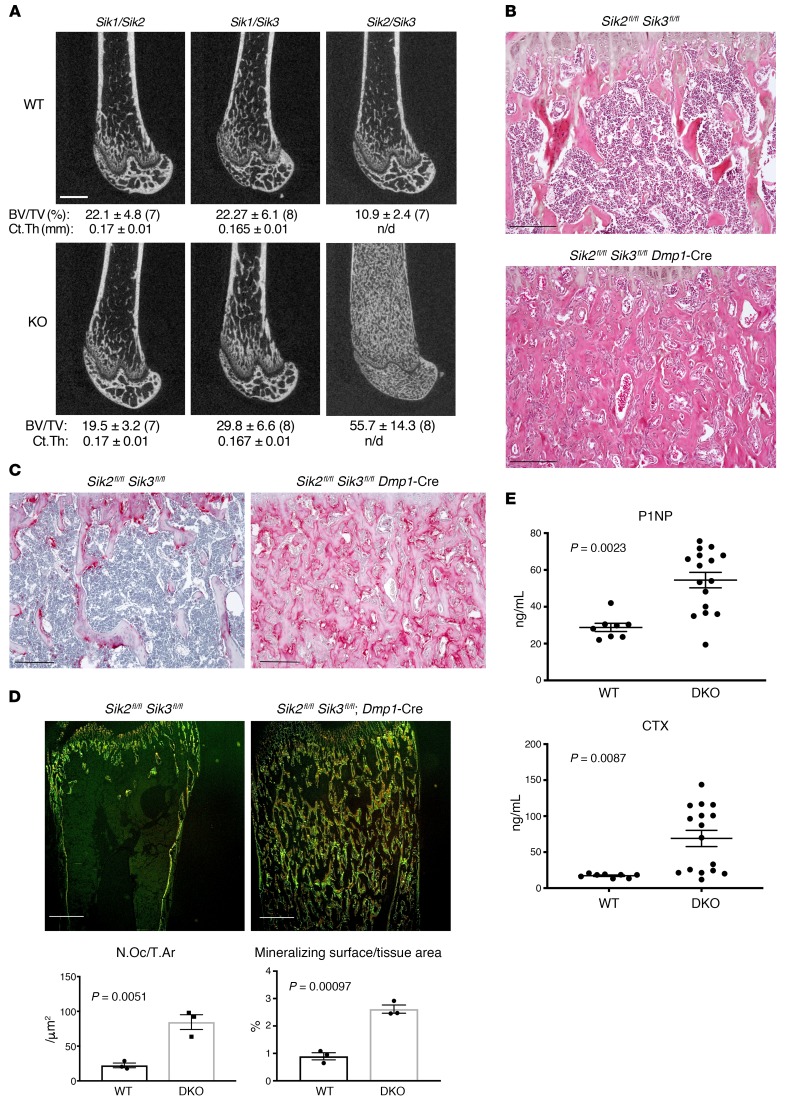
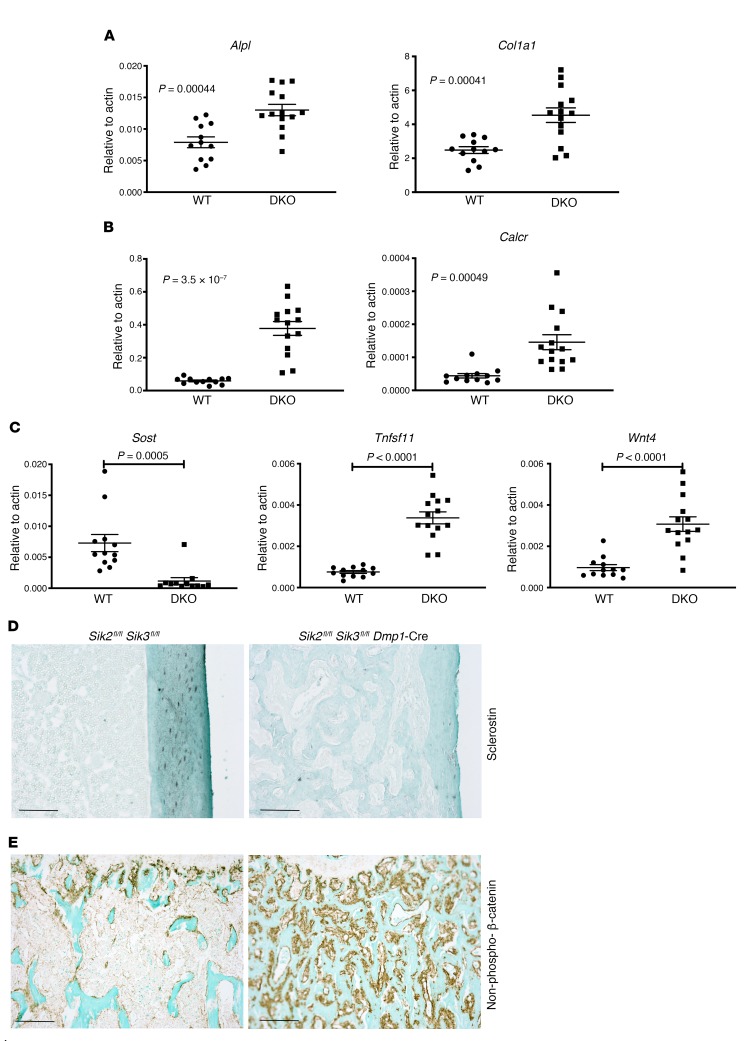
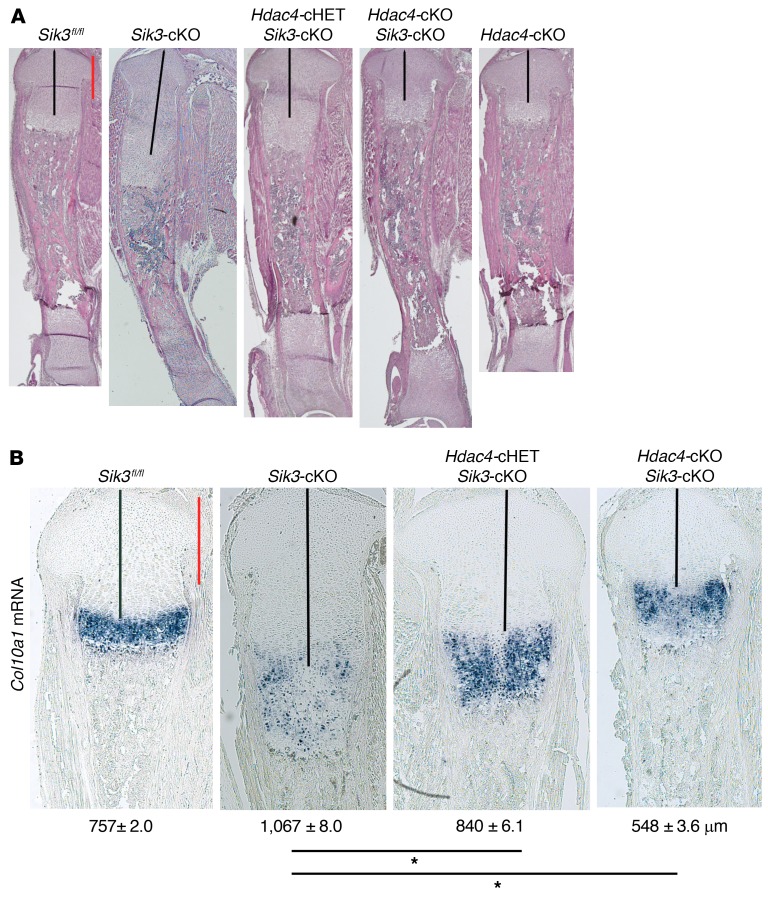
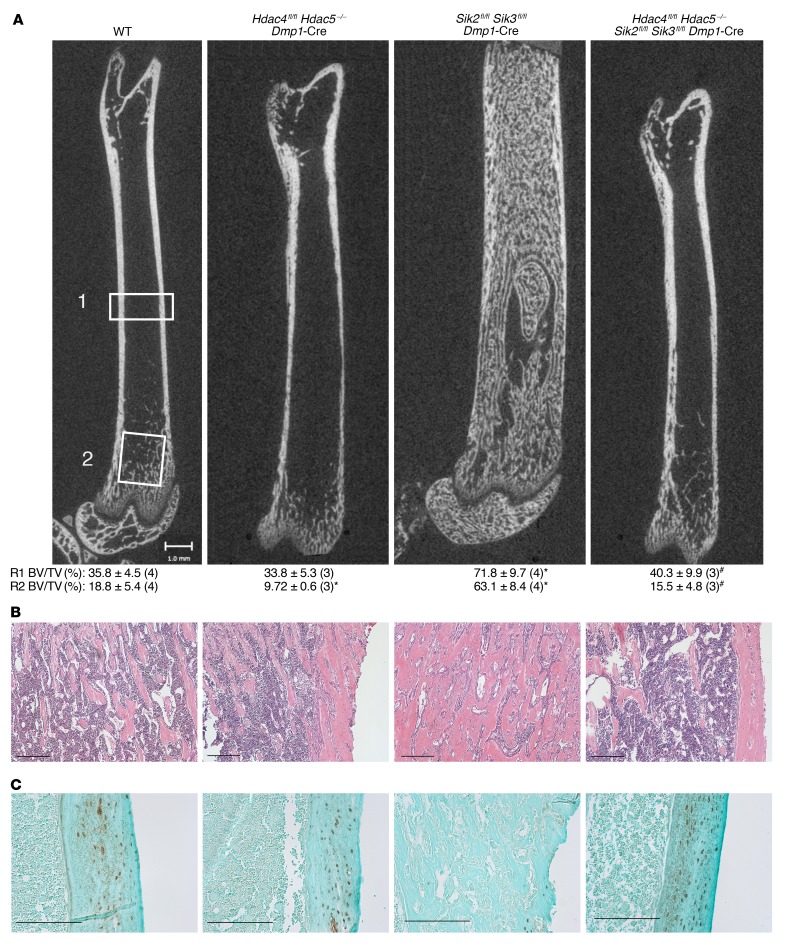
The parathyroid hormone 1 receptor (PTH1R) mediates the biologic actions of parathyroid hormone (PTH) and parathyroid hormone-related protein (PTHrP). Here, we showed that salt-inducible kinases (SIKs) are key kinases that control the skeletal actions downstream of PTH1R and that this GPCR, when activated, inhibited cellular SIK activity. Sik gene deletion led to phenotypic changes that were remarkably similar to models of increased PTH1R signaling. In growth plate chondrocytes, PTHrP inhibited SIK3, and ablation of this kinase in proliferating chondrocytes rescued perinatal lethality of PTHrP-null mice. Combined deletion of Sik2 and Sik3 in osteoblasts and osteocytes led to a dramatic increase in bone mass that closely resembled the skeletal and molecular phenotypes observed when these bone cells express a constitutively active PTH1R that causes Jansen's metaphyseal chondrodysplasia. Finally, genetic evidence demonstrated that class IIa histone deacetylases were key PTH1R-regulated SIK substrates in both chondrocytes and osteocytes. Taken together, our findings establish that SIK inhibition is central to PTH1R action in bone development and remodeling. Furthermore, this work highlights the key role of cAMP-regulated SIKs downstream of GPCR action.
Keywords: Bone Biology; Bone development; Bone disease; Endocrinology; Osteoclast/osteoblast biology.
Conflict of interest statement
Figures


References
-
- Gardella T, Jüppner H, Bringhurst F, Potts J Jr. Receptors for parathyroid hormone (PTH) and PTH-related protein. In: Bilezikian J, Raisz L, Martin T, eds. Principles of Bone Biology. 3rd ed. Amsterdam, Netherlands: Elsevier; 2008:555–576.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
