

Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq)
- PMID: 31431618
- PMCID: PMC6702168
- DOI: 10.1038/s41467-019-11559-1
Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq)
Erratum in
-
Author Correction: Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq).Nat Commun. 2020 Sep 1;11(1):4424. doi: 10.1038/s41467-020-18309-8. Nat Commun. 2020. PMID: 32873803 Free PMC article.
Abstract
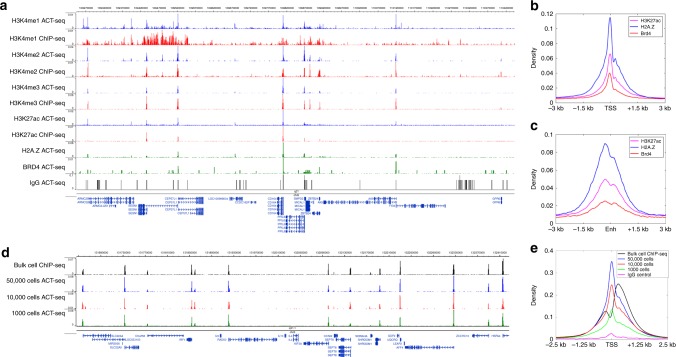
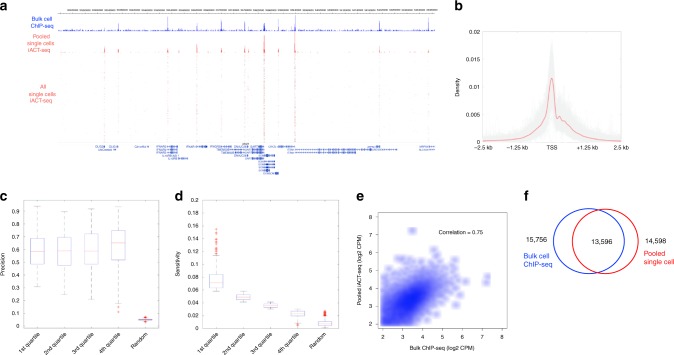
Modern next-generation sequencing-based methods have empowered researchers to assay the epigenetic states of individual cells. Existing techniques for profiling epigenetic marks in single cells often require the use and optimization of time-intensive procedures such as drop fluidics, chromatin fragmentation, and end repair. Here we describe ACT-seq, a streamlined method for mapping genome-wide distributions of histone tail modifications, histone variants, and chromatin-binding proteins in a small number of or single cells. ACT-seq utilizes a fusion of Tn5 transposase to Protein A that is targeted to chromatin by a specific antibody, allowing chromatin fragmentation and sequence tag insertion specifically at genomic sites presenting the relevant antigen. The Tn5 transposase enables the use of an index multiplexing strategy (iACT-seq), which enables construction of thousands of single-cell libraries in one day by a single researcher without the need for drop-based fluidics or visual sorting. We conclude that ACT-seq present an attractive alternative to existing techniques for mapping epigenetic marks in single cells.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
