

The enhancement of glycolysis regulates pancreatic cancer metastasis
- PMID: 31432232
- PMCID: PMC11104916
- DOI: 10.1007/s00018-019-03278-z
The enhancement of glycolysis regulates pancreatic cancer metastasis
Abstract
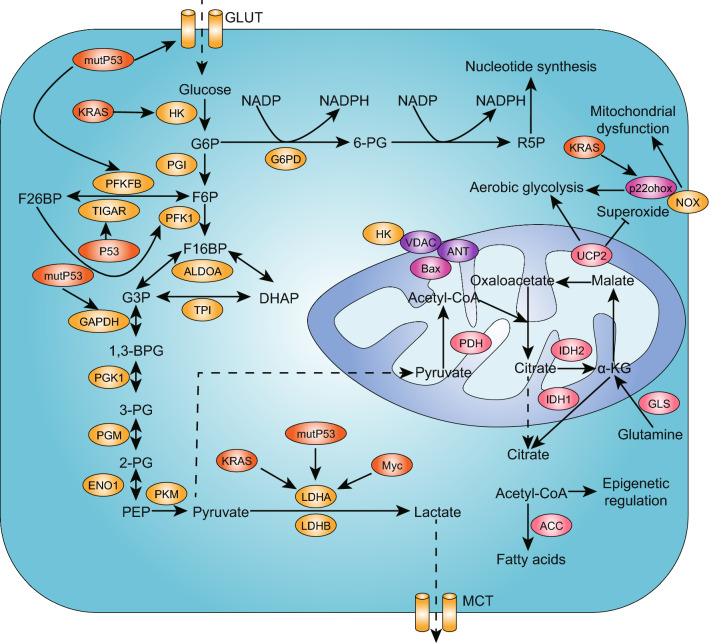
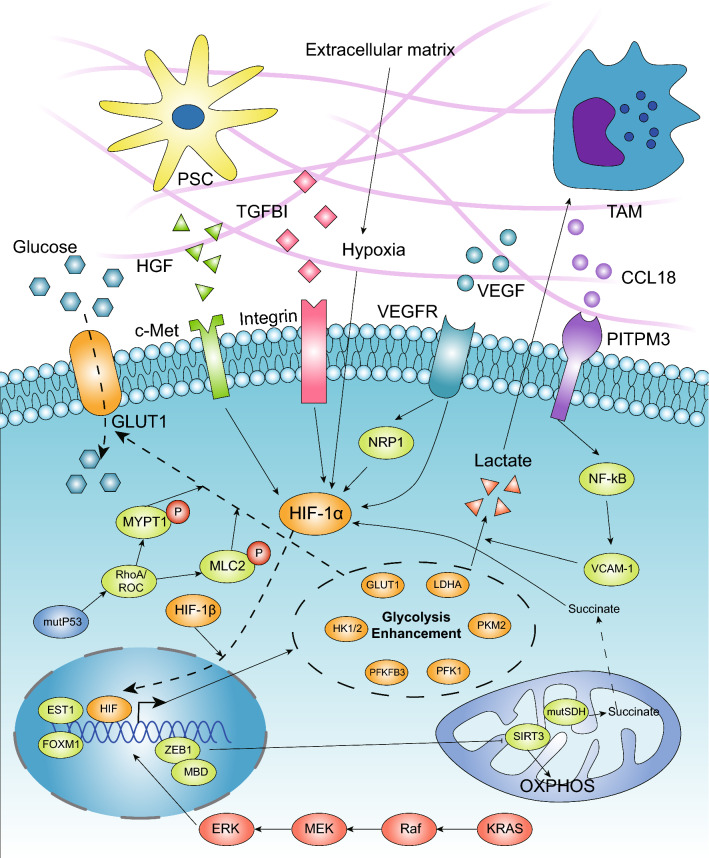
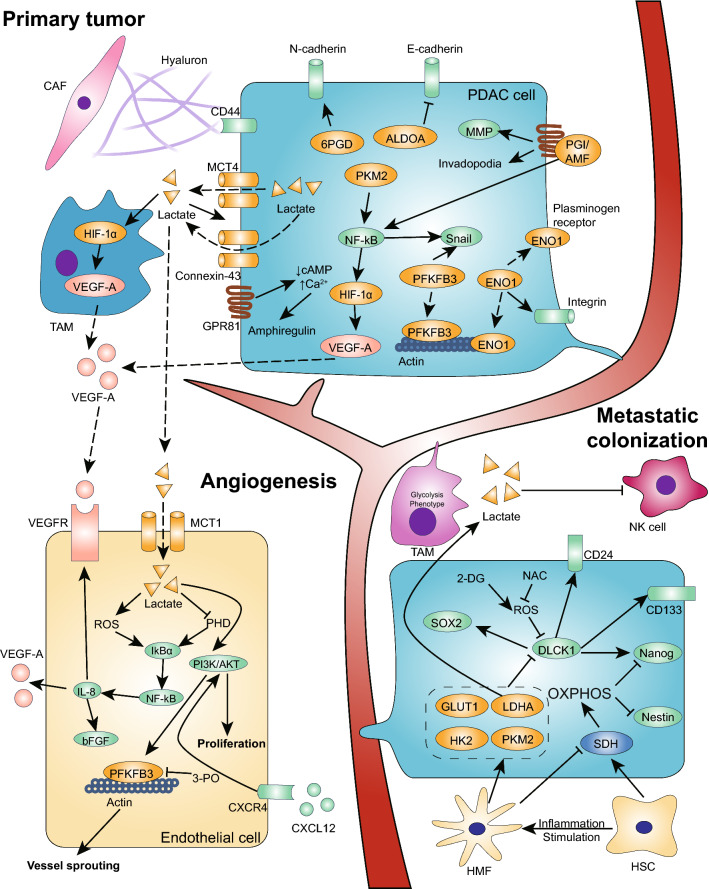
Pancreatic ductal adenocarcinoma is prone to distant metastasis and is expected to become the second leading cause of cancer-related death. In an extremely nutrient-deficient and hypoxic environment resulting from uncontrolled growth, vascular disturbances and desmoplastic reactions, pancreatic cancer cells utilize "metabolic reprogramming" to satisfy their energy demand and support malignant behaviors such as metastasis. Notably, pancreatic cancer cells show extensive enhancement of glycolysis, including glycolytic enzyme overexpression and increased lactate production, and this is caused by mitochondrial dysfunction, cancer driver genes, specific transcription factors, a hypoxic tumor microenvironment and stromal cells, such as cancer-associated fibroblasts and tumor-associated macrophages. The metabolic switch from oxidative phosphorylation to glycolysis in pancreatic cancer cells regulates the invasion-metastasis cascade by promoting epithelial-mesenchymal transition, tumor angiogenesis and the metastatic colonization of distant organs. In addition to aerobic glycolysis, oxidative phosphorylation also plays a critical role in pancreatic cancer metastasis in ways that remain unclear. In this review, we expound on the intracellular and extracellular causes of the enhancement of glycolysis in pancreatic cancer and the strong association between glycolysis and cancer metastasis, which we expect will yield new therapeutic approaches targeting cancer metabolism.
Keywords: Epithelial–mesenchymal transition; Hybrid metabolic phenotype; Metastatic niche; Mitochondrial respiration; Tumor microenvironment; Warburg effect.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Rahib L., Smith B. D., Aizenberg R., Rosenzweig A. B., Fleshman J. M., Matrisian L. M. Projecting Cancer Incidence and Deaths to 2030: The Unexpected Burden of Thyroid, Liver, and Pancreas Cancers in the United States. Cancer Research. 2014;74(11):2913–2921. - PubMed
-
- Giovannetti E, et al. Never let it go: Stopping key mechanisms underlying metastasis to fight pancreatic cancer. Semin Cancer Biol. 2017;44:43–59. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
