

Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease
- PMID: 31432604
- PMCID: PMC6826160
- DOI: 10.1111/acel.13031
Oxidative stress in the aging substantia nigra and the etiology of Parkinson's disease
Abstract
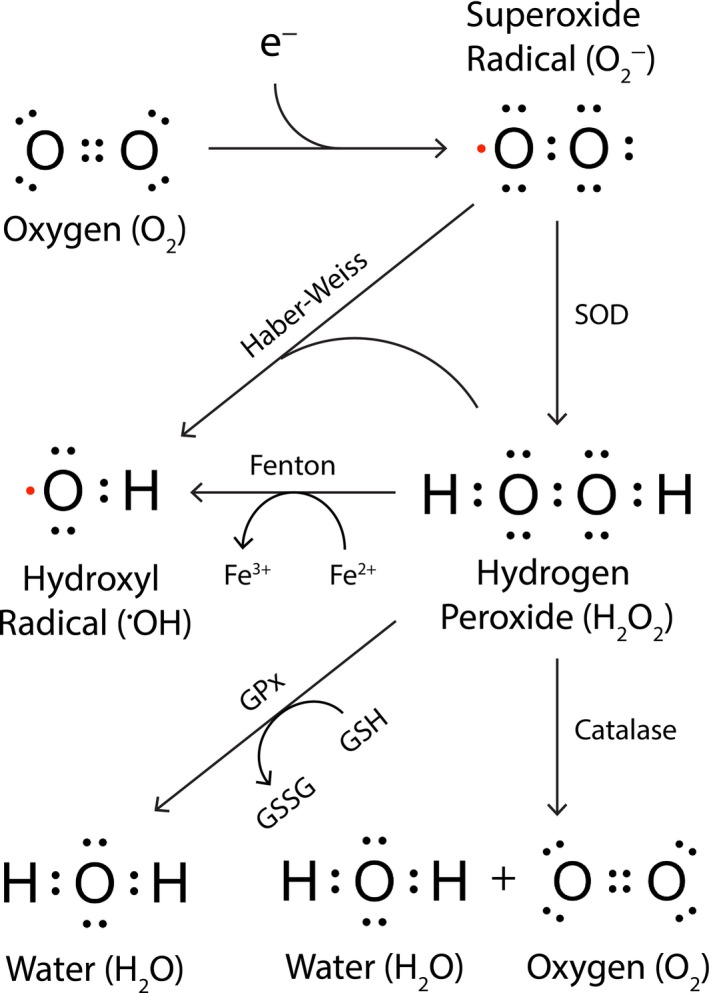
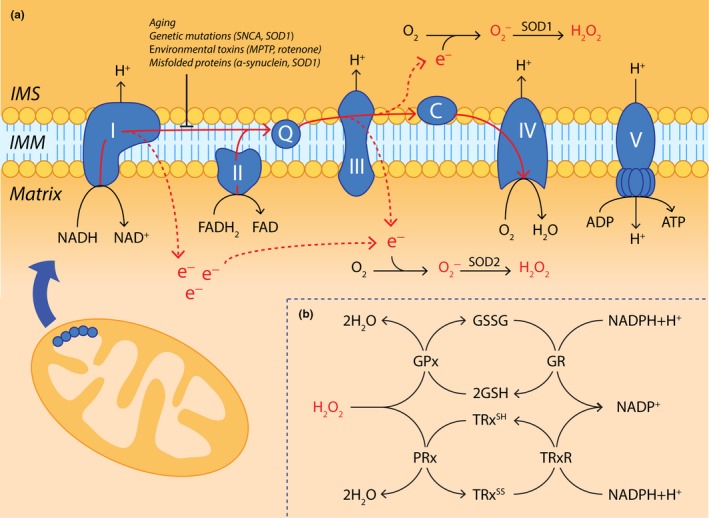
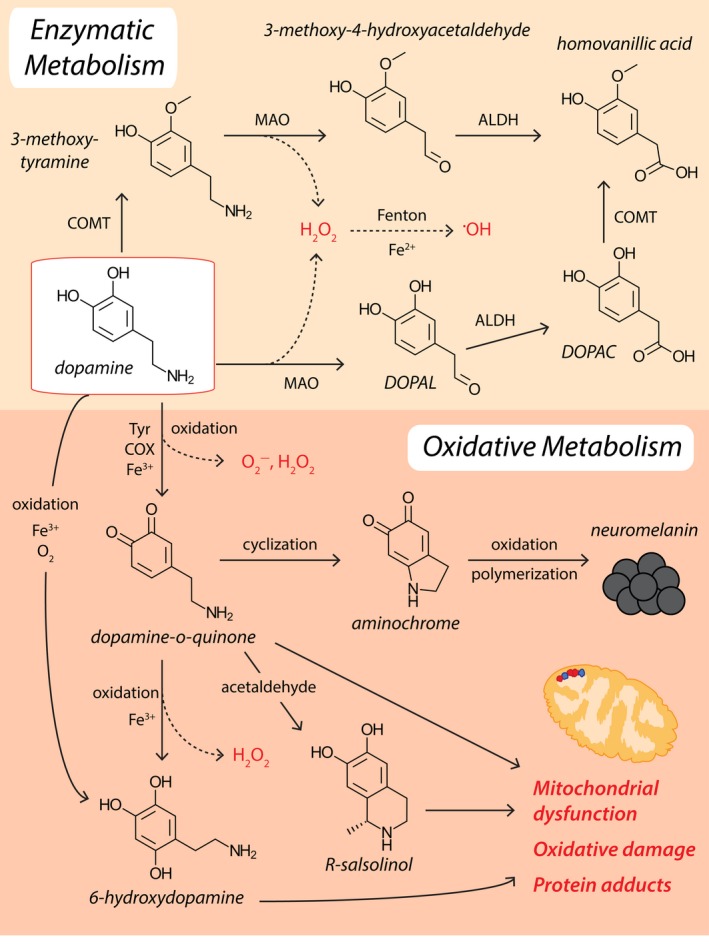
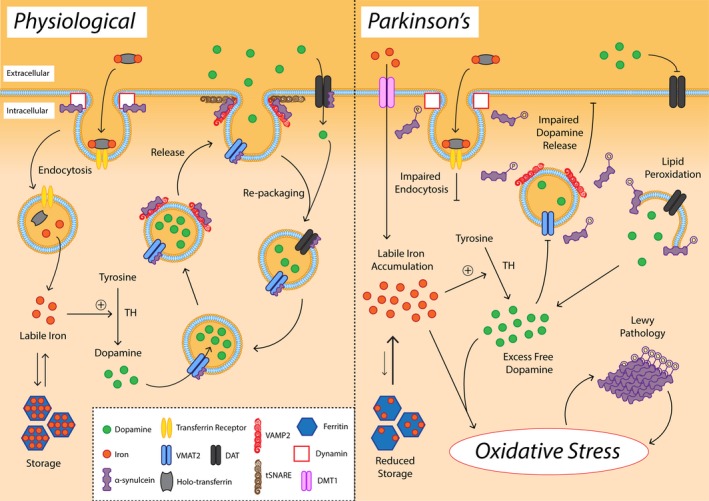
Parkinson's disease prevalence is rapidly increasing in an aging global population. With this increase comes exponentially rising social and economic costs, emphasizing the immediate need for effective disease-modifying treatments. Motor dysfunction results from the loss of dopaminergic neurons in the substantia nigra pars compacta and depletion of dopamine in the nigrostriatal pathway. While a specific biochemical mechanism remains elusive, oxidative stress plays an undeniable role in a complex and progressive neurodegenerative cascade. This review will explore the molecular factors that contribute to the high steady-state of oxidative stress in the healthy substantia nigra during aging, and how this chemical environment renders neurons susceptible to oxidative damage in Parkinson's disease. Contributing factors to oxidative stress during aging and as a pathogenic mechanism for Parkinson's disease will be discussed within the context of how and why therapeutic approaches targeting cellular redox activity in this disorder have, to date, yielded little therapeutic benefit. We present a contemporary perspective on the central biochemical contribution of redox imbalance to Parkinson's disease etiology and argue that improving our ability to accurately measure oxidative stress, dopaminergic neurotransmission and cell death pathways in vivo is crucial for both the development of new therapies and the identification of novel disease biomarkers.
Keywords: Parkinson's disease; antioxidant; oxidative stress; reactive oxygen species.
© 2019 The Authors. Aging Cell published by the Anatomical Society and John Wiley & Sons Ltd.
Conflict of interest statement
None declared.
Figures

References
-
- Ayton, S. , Lei, P. , Hare, D. J. , Duce, J. A. , George, J. L. , Adlard, P. A. , … Bush, A. I. (2015). Parkinson's disease iron deposition caused by nitric oxide‐induced loss of beta‐amyloid precursor protein. Journal of Neuroscience, 35(8), 3591–3597. 10.1523/JNEUROSCI.3439-14.2015 - DOI - PMC - PubMed
-
- Baksi, S. , Tripathi, A. K. , & Singh, N. (2016). Alpha‐synuclein modulates retinal iron homeostasis by facilitating the uptake of transferrin‐bound iron: Implications for visual manifestations of Parkinson's disease. Free Radical Biology and Medicine, 97, 292–306. 10.1016/j.freeradbiomed.2016.06.025 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
